
Abstract
Two Gram-stain-negative, oxidase- and catalase-positive, aerobic strains (CY05T and H18S-6) were isolated from sediment samples of the Yellow Sea, China. The strains were positive for denitrification. Optimum growth was observed at 20 °C, pH 7.5–8.0 and with 2.0%–3.0% NaCl. The predominant cellular fatty acids (> 10%) were summed feature 8 (C18:1 ω7c and/or C18:1 ω6c), major respiratory quinone was ubiquinone-10 and main polar lipids were phosphatidylcholine, phosphatidylglycerol, phosphatidylethanolamine, one unidentified phospholipid and one unidentified aminolipid. The approximate genome size of strains CY05T and H18S-6 were 4.86 and 5.04 Mbp, the genomic G + C content of them were 54.2 and 54.5%, respectively. Both of the phylogenetic analysis based on 16S rRNA gene sequences and the up-to-date bacterial core gene (UBCG) sequences revealed that strains CY05T, H18S-6 and Pelagicola marinus DSW4-44T formed a distinct monophyletic clade within the family Rhodobacteraceae. The ANI and isDDH values between strains CY05T and H18S-6 were 94.0% and 56.5%, between CY05T and Pelagicola marinus DSW4-44T were 94.1% and 59.8%, respectively, all below the accepted threshold value for species delineation. But the ANI and isDDH values between strains H18S-6 and Pelagicola marinus DSW4-44T were 96.8% and 76.7% respectively, indicating that strains H18S-6 and Pelagicola marinus DSW4-44T belong to the same species. Based on the distinctive polyphasic evidence, CY05T represent a novel species of a novel genus of the family Rhodobacteraceae, for which the name Zongyanglinia huanghaiensis gen. nov., sp. nov. is proposed. The type strain is CY05T (= MCCC 1K04409T = KCTC 62200T). Moreover, the reclassification of Pelagicola marinus Choi et al. 2019 as Zongyanglinia marinus comb. nov. (type strain DSW4-44T = KCTC 62762T = KCCM 43261T = JCM 33637T) is proposed based on the polyphasic taxonomic data obtained in this study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The family Rhodobacteraceae is an abundant bacteria group with diverse ecology, metabolism and phenology (Garrity et al. 2005). The number of the genera and species in this family is growing rapidly. For example, 15 novel genera (Park et al. 2018a, b; Feng et al. 2018a, b; Wang et al. 2018; Yu et al. 2018; Klotz et al. 2018; Hu et al. 2018; Zhang et al. 2018; Wirth and Whitman 2018) have been described in 2018, including 6 reclassified novel genera (Cognatishimia, Cognatiyoonia, Flavimaricola, Limimaricola, Pseudaestuariivita and Yoonia) based on phylogenomic analyses (Wirth and Whitman 2018). At the time of writing, the family Rhodobacteraceae contains at least 160 genera encompassing nearly 592 species (List of Prokaryotic Names with Standing in Nomenclature; www.bacterio.net).
Denitrification, an important branch of nitrogen cycle, is a microbial process in which nitrate and nitrite are stepwisely reduced to gaseous forms of nitrogen. As human activities such as the high input of nitrogen-based fertilizers have nearly doubled the nitrogen input to terrestrial and marine ecosystems and consequently lead to water eutrophication, microbial denitrification is getting more attention (Kuypers et al. 2018). Some microbes showing good denitification capacity including Pseudomonas sp. (Sun et al. 2015), Alcaligenes sp. (Ozeki et al. 2001) and Microvirgula sp. (Patureau et al. 2000) have been applied to remove the excess nitrogen from wastewater.
In this study, two bacteria strains showing denitrification capacity were isolated from sediment samples of the Yellow Sea, China. Based on the polyphasic taxonomy study, strain CY05T was proposed to represent a species of a novel genus in the family Rhodobacteraceae. In addition, we explored the existence of genes related to denitrification by analyzing their genomes.
Materials and methods
Samples and cultivation
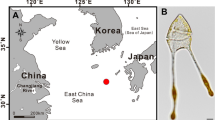
Two surface marine sediment samples were collected at a water depth of 36 m in the Yellow Sea during a cruise as described previously (Zhang et al. 2017) in 2014. Samples were stored in sterilized plastic bags (250 mL) and transported to the laboratory at 15 ºC. Bacterial strains were isolation using the standard dilution plating technique on TYM agar at 15ºC as described previously (Zhang et al. 2018). Strain CY05T was isolated from sample collected at the site H02 (36° N, 121° E) in November, and H18S-6 was isolated from sample collected at the site H18 (35° N, 121° E) in May, respectively. The temperature and salinity of the seawater at site H02 and H18 measured by CTD system was 18.2ºC and 3.1%, 10.1ºC and 3.1%, respectively. In addition, the following strains: Pelagicola marinus DSW4-44T (obtained from Korean Collection for Type Cultures), Pelagicola litoralis DSM 18290T, Phaeobacter porticola DSM 103148T, Sulfitobacter pseudonitzschiae MCCC 1A00686T, Sedimentitalea nanhaiensis MCCC 1A04178T and Pelagimonas varians DSM 23678T (obtained from Marine Culture Collection of China) were characterized alongside for comparative purposes. Unless otherwise stated, these strains were routinely cultured in marine broth 2216 (MB; Difco) or on marine agar 2216 (MA; MB with 1.5% agar) at 20ºC except for Pelagimonas varians DSM 23678T cultured in corresponding medium supplemented with 0.25 mg/L nicotinic acid.
Phylogenetic analysis
The 16S rRNA gene was amplified by PCR with the universal primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) (Weisburg et al. 1991). PCR products were purified using the GeneJET gel extraction kit (Thermo). The purified PCR products were ligated into pMD 19-T vector (Tsingke) and sequenced using an automated DNA sequencer (model 3730xl; Applied Biosystems) at Tsingke Biological Technology Co. Ltd (Qingdao, China). The nearly complete 16S rRNA gene sequences were compared to those of species validly published through the EzBioCloud server [http://www.ezbiocloud.net/, (Yoon et al. 2017)]. Phylogenetic trees were constructed with MEGA 6 (Tamura et al. 2013) using maximum-likelihood (Felsenstein 1981), neighbour-joining (Saitou and Nei 1987) and maximum-parsimony (Fitch 1971) methods. The topologies of the phylogenetic trees were evaluated by bootstrap analyses (1000 replications).
Genomic DNA sequencing and analyses
Genomic DNA of strains CY05T and H18S-6 were extracted using a bacterial genomic DNA isolation kit (BioTeke) following the manufacturer’s instructions except that sterile ddH2O was used for DNA elution. Genome sequencing was performed at BGI-Shenzhen (China) using the HiSeq 4000 sequencer system (Illumina). The DNA G + C % content of the two strains were calculated directly from the draft genome sequences.
The obtained draft genomes were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al. 2016) and RAST v2.0 (Rapid Annotation using Subsystem Technology) (Brettin et al. 2015).
Phylogenomic tree reconstruction and genome comparison
Phylogenomic tree reconstruction was conducted based on an up-to-date bacterial core gene set (UBCG) consisting of 92 single-copy core genes (Na et al. 2018). The alignment file of the UBCG was generated using a JAVA program (Na et al. 2018) from the genome sequences and was used as input for the PhyML 3.0 server (www.atgc-montpellier.fr/phyml/) with smart model selection (Guindon et al. 2010; Lefort et al. 2017). Robustness estimation of the nodes is conducted using bootstrapping with 100 replicates. Genome-based similarity indexes including in silico DNA-DNA hybridization (isDDH), average nucleotide identities (ANI), average amino acid identities (AAI) and the percentage of conserved proteins (POCP) were also calculated. The isDDH values were estimated with the Genome-to-Genome Distance Calculator using the recommended formula 2. ANI values including ANIm and ANIb were calculated in JspeciesWS. AAI values were determined by the website-based AAI calculator (Rodriguez-R and Konstantinidis 2016). POCP values were calculated according to Qin et al. (2014).
Morphological, physiological and biochemical analysis
The colony morphologies of the strains were observed after incubation on MA at 20 ºC for 7 days. Cellular morphologies and flagellum were observed by transmission electron microscope (HITACHI HT7700) with cells from exponentially growing cultures. Flagellum was also checked by the flagella staining method (Qingdao Hope Bio-Technology Co. Ltd., China). Gram staining was carried out using the standard Gram procedure (Murray et al. 1994). Growth at temperatures (5, 10, 15, 20, 25, 30, 37 and 42 °C) was measured in MB. Growth with different NaCl concentrations (0, 0.5% and 1.0%–10.0%, at intervals of 1.0% units; w/v) was determined in NP broth (Zhang et al. 2017) at 20 °C. Growth tests for pH range [5.0–10.0, at intervals of 0.5 pH units, buffered with MES (pH 5.0–6.0, 50 mM), MOPS (pH 6.5–7.0, 50 mM), Tris (pH 7.5–8.5, 50 mM) and CHES (pH 9.0–10.0, 50 mM)] were assessed in NP broth with 2.0% NaCl. Oxidase activity was tested by using commercial oxidase test strips (Tianhe Microorganism Reagent Co.) according to the manufacturer’s instructions. Catalase activity was determined by bubble production in 3% (v/v) H2O2. Hydrolysis of starch, casein, Tween 40, Tween 60 and Tween 80 were examined by using MA as the basal medium and incubation at 20 °C. Growth under anaerobic condition was determined in MB supplemented with potassium nitrate (0.1%, w/v), cysteine hydrochloride (0.05%, w/v) and sodium sulfide (0.05%, w/v) in Hungate tubes filled with oxygen-free N2 at 20ºC for two weeks. The utilization of carbohydrates (0.5%, w/v) as sole carbon and energy sources was determined in basal medium (Farmer et al. 2005) at 20 °C for 14 days. Growth was scored as negative when it was less than, or equal to, that in the negative control (lacking any carbon source). Nitrate reduction, denitrification and nitrite reduction were tested in MB medium supplemented with KNO3 (0.1%, w/v) and NaNO2 (0.01%, w/v) respectively, and contained a small inverted vial. The result were examined as (Dong and Cai 2001) described. MB medium contained a small inverted vial served as control. Susceptibility to antibiotics was examined by the disc-diffusion method on MA after spreading cell suspensions (0.5 McFarland). The discs (Oxoid) contained the following antibiotics: amoxycillin (10 μg), ampicillin (10 μg), bacitracin (10 U), carbenicillin (100 μg), cefotaxime (30 μg), cefoxitin (30 μg), chloramphenicol (30 μg), nitrofurantoin (300 μg), penicillin G (10 U) and rifampicin (5 μg). Antibiotic susceptibility was assessed by measuring diameters of the inhibition zone after 5 days at 20 °C. An inhibition zone diameter more than 10 mm (including the 6 mm diameter of the filter paper) was considered sensitive to antimicrobial agent (Wu et al. 2015). More biochemical properties were determined by using API ZYM and API 20NE strips (bioMerieux) according to the manufacturer’s instructions with sterilized artificial seawater (ASW) (Tindall et al. 2007) as the cell suspension solution and AUX medium supplemented with 2.0% (w/v) NaCl. The results were read after incubation at 20 °C for 24 h, 24 and 48 h for API ZYM and API 20 NE kits, respectively.
Chemotaxonomic analysis
For the analysis of cellular fatty acids, cells were collected from the third quadrant of the quadrant streaked plate (at exponential phase) after incubated on MA medium supplemented with 0.25 mg/L nicotinic acid at 20ºC. Fatty acid methyl esters were analysed by an Agilent 6850 N gas chromatograph and identified using the Sherlock Microbial Identification System (TSBA, version 6.1, MIDI) at Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao, P. R. China. For the analysis of polar lipids and quinone, cells of them were grown to early stationary phase in MB at 20 °C. Polar lipids were extracted according to (Komagata and Suzuki 1988) and analysed using two-dimensional TLC on silica gel 60 F254 plates (Merck) with appropriate spraying reagents including ethanolic molybdophosphoric acid (total lipids), ninhydrin (aminolipids) and molybdenum blue (phospholipids). The respiratory quinone of strains CY05T and H18S-6 were extracted, separated and analysed as described by (Lin et al. 2015).
Results and discussion
Phylogenetic analysis
Nearly full-length 16S rRNA gene sequence of CY05T (1425 bp, MN790778) and H18S-6 (1425 bp, MN808553) were obtained. Strains CY05T and H18S-6 showed the highest 16S rRNA gene sequence similarities to Pelagicola marinus DSW4-44T (100% and 99.93%) and less than 96.6% sequence similarities with the closely related type strains of the family Rhodobacteraceae in the class Alphaproteobacteria. The 16S rRNA gene sequence similarity between CY05T and H18S-6 was 99.93%. There was also relatively similar sequence similarities of strains CY05T and H18S-6 to closely related species Pelagicola litoralis DSM 18290T (96.54% and 96.47%, respectively), Phaeobacter porticola DSM 103148T (96.32% and 96.32%, respectively), Sulfitobacter pseudonitzschiae MCCC 1A00686T (96.17% and 96.10%, respectively), Sedimentitalea nanhaiensis MCCC 1A04178T (95.96% and 95.89%, respectively) and Pelagimonas varians DSM 23678T (95.75% and 95.68%, respectively). In the maximum-likelihood tree (Fig. 1), strains CY05T, H18S-6 and Pelagicola marinus DSW4-44T formed a monophyletic clade within the family Rhodobacteraceae with 100% bootstrap support. This stable topology was also supported by the maximum-parsimony (Fig. S1) and neighbour-joining trees (Fig. S2). On the basis of their stable monophyletic grouping, strains CY05T, H18S-6 and Pelagicola marinus DSW4-44T should be placed in a new genus of the family Rhodobacteraceae.

Maximum-likelihood phylogenetic tree based on 16S rRNA gene sequences of strains CY05T, H18S-6 (in bold) and members of closely related taxa. Hyphomonas polymorpha PS728T was used as an outgroup. Bootstrap values (> 70%) based on 1000 replicates are shown at branch points. Bar, 0.02 substitutions per nucleotide position
Phylogenomic tree reconstruction and genome comparison
Previous studies had shown that 16S rRNA gene sequences lack the resolution for a proper phylogenetic reconstruction inside the Rosebacter group (Luo and Moran 2014; Wirth and Whitman 2018) and genome sequence data was strongly recommended to use for the taxonomy of these bacteria. Therefore, a multigene, genome-based tree using the UBCG tool was constructed to complement the 16S rRNA gene-based trees. In accordance with the 16S rRNA trees, the genome-based tree (Fig. 2) also showed that strains CY05T, H18S-6 and Pelagicola marinus DSW4-44T formed a distinct phylogenetic clade (100% bootstrap value support) within the family Rhodobacteriaceae conforming that strains CY05T, H18S-6 and DSW4-44T should be considered as representing a novel genus of the family Rhodobacteraceae. Further, genome-based similarity indexes including isDDH, ANI, AAI and POCP which were useful for the taxonomy of prokaryotes were calculated and depicted in Table 1. ANI and isDDH percentages were genomic nucleic acid-level comparisons widely used for species delineation. As shown in Table 1, ANIb percentages between strains CY05T and H18S-6, Pelagicola marinus DSW4-44T, Pelagicola litoralis DSM 18290T, Phaeobacter porticola DSM 103148T, Sulfitobacter pseudonitzschiae MCCC 1A00686T, Sedimentitalea nanhaiensis MCCC 1A04178T and Pelagimonas varians DSM 23678T were 94.0%, 94.1%, 69.7%, 72.2%, 69.5%, 70.6% and 68.7%, respectively, and the isDDH values between them were 56.5%, 59.8%, 20.0%, 19.5%, 19.6%, 17.7% and 19.7%, respectively. They were all below the threshold value for ANI (95%–96%) and isDDH (70%) to discriminate bacterial species (Chun et al. 2018). ANI and isDDH percentages indicated that strains CY05T represents a novel genomic species in the family Rhodobacteraceae. The isDDH and ANIb values were 76.7% and 96.8% after comparing strains H18S-6 and Pelagicola marinus DSW4-44T. Considering the recommended threshold value for species discrimination (isDDH < 70% and ANI < 95%–96%), it is clear that strains H18S-6 and Pelagicola marinus DSW4-44T belong to the same species based on the high values of isDDH and ANIb values between them. AAI and POCP percentages were genomic amino acid-level comparisons mainly used for genus delineation. Previously studies showed that the AAI percentages worked perfectly for delimiting genera in the Roseobacter group (Wirth and Whitman 2018) by a gradient of AAI percentages defined by two values: a minimum value (80%) below which species should be separated into different genera. Otherwise, the suggested genus boundary of 50% POCP was found to be too conservative and consequently it was inappropriate for delimiting genera in the Roseobacter group (Wirth and Whitman 2018) and many other bacterial genera (Aliyu et al. 2016; Li et al. 2017). As shown in Table 1, AAI percentages among CY05T, H18S-6 and Pelagicola marinus DSW4-44T were 95.8–97.9%, which were significantly higher than the threshold value for AAI (> 80%) to discriminate bacterial genera (Wirth and Whitman 2018). This result indicated that they represented a genus-level taxon in agreement with the result of the 16S rRNA gene phylogeny, phylogenomic tree and the genome comparison, including ANI and isDDH values. For the POCP percentages, our results confirmed that 50% POCP alone could not be applied to Roseobacter group for genus-level circumscription because all of the calculated POCP percentages were greater than this value. All together, the genome sequence data based on ANI, isDDH and AAI values indicated that strains CY05T, H18S-6 and Pelagicola marinus DSW4-44T represent a novel genus of the family Rhodobacteraceae.

UBCG maximum-likelihood tree. Tree was generated using PhyML server 3.0 with smart model selection. The numbers at the nodes indicate the bootstrap support after 100 replicates. Hyphomonas polymorpha PS728T was used as an outgroup. Bar, 0.1 substitutions per position
Genomic DNA sequencing and analyses
The assembled draft genome of strain CY05T was 4,856,535 bp in length, with 40 contigs and a N50 value of 277,157 bp. According to annotation by PGAP, A total of 4660 genes were predicted, with 4557 protein-coding genes, 53 RNA genes and 50 pseudogenes. The assembled draft genome of strain H18S-6 was 5,037,133 bp in length, with 292 contigs and a N50 value of 264,929 bp. According to annotation by PGAP, A total of 4934 genes were predicted, with 4808 protein-coding genes, 69 RNA genes and 57 pseudogenes. The calculated genomic DNA G + C content of strains CY05T and H18S-6 were 54.5 and 54.2%, similar to that of Pelagicola marinus DSW4-44T (54.3%), but relatively lower than those of other type strains in related genera (Table 2).
In accordance with the phenotypic results that strains CY05T and H18S-6 could reduce nitrate to gaseous forms of nitrogen, the genomes of strains CY05T and H18S-6 contained a complete set of genes encoding the enzymes involved in denitrification, including nitratereductase, nitrite reductase, nitric oxide reductase and nitrous-oxide reductase (Table. S1). Although the related genes of denitrification of the two isolates are located in different positions far away from each other, suggesting that these genes may not function simultaneously, strains CY05T and H18S-6 were positive for nitrate and nitrite reduction and denitrification based on the experimental results. There may be other ways to compensate and the specific reasons need to be further explored. Furthermore, based on the genome annotation and gene sequence comparison, the genome of strain H18S-6 also contained an assimilatory pathway by which this strain could reduce nitrate to ammonia for growth (Table. S2). However, whether or not these genes could function is not clear.
Morphological, physiological and biochemical analysis
Both strains were Gram negative, aerobic, rod-shaped and non-flagellated. Growth of the two strains was found to occur between 4 and 30 °C (optimum 20 °C). They were able to reduce nitrate and nitrite to gaseous forms of nitrogen. They were sensitive to amoxycillin (10 μg), ampicillin (10 μg), bacitracin (10 U), carbenicillin (100 μg), cefotaxime (30 μg), cefoxitin (30 μg), chloramphenicol (30 μg), nitrofurantoin (300 μg), penicillin G (10 U) and rifampicin (5 μg). There were many differences between the two strains. Cells of strains CY05T and H18S-6 were (0.7–1.8) µm and (0.8–1.3) µm in width, and (1.6–3.9) µm and (1.9–4.5) µm in length, respectively (Fig. S3). Growth occurs in 1.0%–5.0% NaCl (optimum, 2.0%–3.0%) for strain CY05T and 2.0–5.0% (optimum, 3.0%) for strain H18S-6. The pH for growth was pH 6.5–9.0 (optimum, 7.5–8.0) for CY05T and pH 7.0–9.0 (optimum, 7.5) for H18S-6. The strain H18S-6 also distinguished from strain CY05T by the ability to utilise D-cellobiose and inositol as single carbon resource for growth and the presence of α-galactosidase activities. The strains CY05T differed from those of phylogenetically related taxa by the ability of nitrate reduction and denitrification, flagellum, the assimilation of carbohydrates, the tolerance to sodium chloride and temperatures. (Table 2 and S3). Further data on the morphological, physiological and biochemical characteristics of the isolates were mentioned in Table 2 and the species description.
Chemotaxonomic analysis
The chemotaxonomic results also supported the results of the phylogenetic analysis. Cellular fatty acid comparison of CY05T, H18S-6 and the type strains of closely related species were shown in Table 3. The predominant cellular fatty acids of strains CY05T and H18S-6 were summed feature 8 (C18:1 ω7c and/or C18:1 ω6c) (> 80%), which are characteristics of the family Rhodobacteraceae. But the proportions of some fatty acids were distinguishable. C16:0 were the significant amounts in strains Pelagicola litoralis DSM 18290T, Phaeobacter porticola DSM 103148T, Sulfitobacter pseudonitzschiae MCCC 1A00686T and Pelagimonas varians DSM 23678T, but were detected at lower levels in strains CY05T, H18S-6, Pelagicola marinus DSW4-44T and Sedimentitalea nanhaiensis MCCC 1A04178T. Strain Sedimentitalea nanhaiensis MCCC 1A04178T contained a higher amount of C12:0 3OH, but it was not detected in CY05T, H18S-6, Pelagicola marinus DSW4-44T (Table 3). The distinctiveness of our isolates was also evident in the polar lipid profiles. The polar lipids of strain CY05T included phosphatidylcholine, phosphatidylglycerol, phosphatidylethanolamine, one unidentified aminolipid, three unidentified phospholipids, one unidentified lipid and one unidentified amino phospholipid. The polar lipids of strain H18S-6 included phosphatidylcholine, phosphatidylglycerol, phosphatidylethanolamine, three unidentified phospholipids, two unidentified aminolipids and one unidentified lipid. Unidentified amino phospholipid was not detected in other strains except for strains CY05T and Pelagimonas varians DSM 23678T. However, more unidentified lipids were detected in Pelagimonas varians DSM 23678T than in strain CY05T. Two unidentified aminolipids were only detected in strain H18S-6 (Fig. S4). The major respiratory quinone detected in strains CY05T and H18S-6 was ubiquinone-10, typical in members of the family Rhodobacteraceae (Park and Yoon 2013).
In conclusion, strain CY05T is not closely affiliated with any closely related genera by polyphasic characteristic differences, such as the ability of nitrate reduction, nitrite reduction and denitrification, the tolerance to sodium chloride and temperatures (Table 2). Especially significant differences between the content of polar lipids, fatty acid profiles and DNA G + C contents as described above. Furthermore, the phylogenetic position based on the 16S rRNA gene sequences and the genome sequence also reflected this relationship. And strain CY05T could also be distinguished from H18S-6 to be recognized as separate species, such as culture conditions, the ability to utilise D-cellobiose and inositolthe, content of polar lipids and fatty acid profiles (Table 2).
Based on the polyphasic data presented in this study, strain CY05T should be considered to represent a novel species of a novel genus within the family Rhodobacteraceae, for which the name Zongyanglinia huanghaiensis gen. nov. sp. nov. is proposed. Moreover, the genome sequence data based on ANI, isDDH and AAI values indicated that strains H18S-6 and Pelagicola marinus DSW4-44T belong to the same species. Combined with the above mentioned polyphasic data, such as the phylogenetic trees and the genome sequence data based on ANI, isDDH and AAI values, strain DSW4-44T should be transferred to this new genus Zongyanglinia as Zongyanglinia marinus comb. nov., the type strain is DSW4-44T (= KCTC 62762T = KCCM 43261T = JCM 33637T).
Description of Zongyanglinia gen. nov.
Zongyanglinia (Zong.yang.li’ni.a. N.L. fem. N. Zongyanglinia named to honour Chinses microbiologist Zong-Yang Lin).
Cells are Gram-stain negative. The major respiratory quinone is ubiquinone-10 (Q-10). The main polar lipids included phosphatidylcholine, phosphatidylglycerol, phosphatidylethanolamine, unidentified phospholipid, unidentified aminolipid and lipid. The predominant cellular fatty acids are summed feature 8 (C18:1 ω7c and/or C18:1 ω6c). The DNA G + C content of the genomic DNA are 54.2%–54.3%. Phylogenetically, the genus Zongyanglinia is affiliated to the family Rhodobacteraceae of the class Alphaproteobacteria. The type species is Zongyanglinia huanghaiensis CY05T (= MCCC 1K04409T = KCTC 62200T).
Description of Zongyanglinia huanghaiensis sp. nov.
Zongyanglinia huanghaiensis (hu.ang.hai.en’sis. N.L. masc. adj. huanghaiensis pertaining to Huanghai, the Chinese name for the Yellow Sea, the geographical origin of the type strain).
Strain exhibits the following properties in addition to those given in the genus description. Cells are aerobic, rods or ovoid-shaped rods and approximately 0.7 μm –1.8 μm in width and 1.6 μm –3.9 μm in length. Colonies are circular with regular edges, opaque, slightly convex and approximately 0.2 mm–1.1 mm in diameter on MA agar after 7 days incubation at 20 °C. Growth occurs at 5 °C–30 °C (optimum, 20 °C) and in pH range 6.5–9 (optimum, 7.5–8.0). Grows with 1.0%–5.0% NaCl (optimum, 2.0%–3.0%), does not grow without NaCl. Positive for catalase, oxidase, nitrate and nitrite reduction and denitrification, but could not hydrolysis of casein, starch, Tween 40, Tween 60 and Tween 80. The following substances are utilised as single carbon resource for growth: D-glucose, sucrose, inulin, D-raffinose pentahydrate, D-galactose, D-mannose, D-maltose monohydrate and xylan (weakly), but not D-cellobiose, inositol, D-xylose, xylitol, D-fructose, D-mannitol, sorbitolum, L-sorbose, L-arabinose and D-trehalose. In the API ZYM strips, positive for alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, valine arylamidase, acid phosphatase and naphthol-AS-B1-phosphophydrolase, but negative for lipase (C14), cystine arylamidase, trypsin, α-chymotrypsin, α-galactosidase, β-galactosidase, β-glucuronidase, α-glucosidase, β-glucosidase, N-acetyl-β-glucosaminidase, α-mannosidase and α-fucosidase activities. In API 20NE tests, positive for nitrate reduction, but negative for indole production, acid production from glucose, arginine dihydrolase, urease, β-glucosidase, gelatinase and beta-galactosidase, or assimilattion of glucose, arabinose, mannose, mannitol, N-acetyl-glucosamine, maltose, gluconate, caprate, adipate, malate, citrate and phenyl-acetate.
The type strain is CY05T (= MCCC 1K04409T = KCTC 62200T), isolated from sediment of the Yellow Sea. The G + C content of the genome is 54.2%, its approximate genome size is 4.86 Mbp. The GenBank accession numbers for the 16S rRNA gene sequence and the draft genome sequence of the type strain are MN790778 and WQLV00000000, respectively.
Description of Zongyanglinia marinus comb. nov.
Zongyanglinia marinus (ma.ri′nus. L. masc. adj. marinus of the sea, marine).
Basonym: Pelagicola marinus Choi et al. 2019.
The description is the same as that given by Choi et al. (2019) with the following modification. Positive for nitrite reduction and denitrification, could grow at 5ºC, weakly utilize inulin as single carbon resource for growth. The type strain is DSW4-44T (= KCTC 62762T = KCCM 43261T = JCM 33637T).
References
Aliyu H, Lebre P, Blom J, Cowan D, De Maayer P (2016) Phylogenomic re-assessment of the thermophilic genus Geobacillus. Syst Appl Microbiol 39:527–533. https://doi.org/10.1016/j.syapm.2016.09.004
Breider S, Freese HM, Spröer C, Simon M, Overmann J, Brinkhoff T (2017) Phaeobacter porticola sp. nov., an antibiotic-producing bacterium isolated from a sea harbour. Int J Syst Evol Microbiol 67:2153–2159. https://doi.org/10.1099/ijsem.0.001879
Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ et al (2015) RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep 5:8365. https://doi.org/10.1038/srep08365
Choi YS, Oh JS, Roh DH (2019) Pelagicola marinus sp nov isolated from deep-sea water. Int J Syst Evol Microbiol 69(12):3961–3966. https://doi.org/10.1099/ijsem.0.003764
Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS, Rooney AP, Yi H, Xu XW, De Meyer S, Trujillo ME (2018) Proposed minimal standards for the use of genome data forthe taxonomy of prokaryotes. Int J Syst Evol Microbiol 68(1):461–466. https://doi.org/10.1099/ijsem.0.002516
Dong X, Cai M (2001) Manual for the systematic identification of general bacteria Beijing. Science Press, Beijing
Farmer IIIJ, Janda JM, Brenner FW, Cameron DN, Birkhead KM et al (2005) Genus I. Vibrio Pacini 1854, 411AL. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds) Bergey’s Manual of Systematic Bacteriology, vol 2, 2nd edn. The Proteobacteria, Part B, The Gammaproteobacteria, New York, p 494
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376. https://doi.org/10.1007/BF01734359
Feng T, Jeong SE, Kim KH, Park HY, Jeon CO (2018a) Aestuariicoccus marinus gen. nov., sp. nov., isolated from sea-tidal flat sediment. Int J Syst Evol Microbiol 68:260–265. https://doi.org/10.1099/ijsem.0.002494
Feng T, Kim KH, Jeong SE, Kim W, Jeon CO (2018b) Aquicoccus porphyridii gen. nov., sp. nov., isolated from a small marine red alga, Porphyridium marinum. Int J Syst Evol Microbiol 68:283–288. https://doi.org/10.1099/ijsem.0.002498
Fitch WM (1971) Toward defining the course of evolution: minimum change for a specific tree topology. Syst Biol 20:406–416. https://doi.org/10.2307/2412116
Garrity G, Bell J, Lilburn T, Family I (2005) Rhodobacteraceae fam nov. In: Garrity GM (ed) Bergey’s manual of systematic bacteriology. Springer, US
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. https://doi.org/10.1093/sysbio/syq010
Hahnke S, Tindall BJ, Schumann P, Simon M, Brinkhoff T (2013) Pelagimonas varians gen. nov., sp. nov., isolated from the southern North Sea. Int J Syst Evol Microbiol 63:835–843. https://doi.org/10.1099/ijs.0.040675-0
Hong Z, Lai Q, Luo Q, Jiang S, Zhu R, Liang J, Gao Y (2015) Sulfitobacter pseudonitzschiae sp. nov., isolated from the toxic marine diatom Pseudo-nitzschia multiseries. Int J Syst Evol Microbiol 65:95–100. https://doi.org/10.1099/ijs.0.064972-0
Hu Q, Zhang L, Hang P, Zhou XY, Jia WB, Li SP, Jiang JD (2018) Xinfangfangia soli gen. nov., sp. nov., isolated from a diuron-polluted soil. Int J Syst Evol Microbiol 68:2622–2626. https://doi.org/10.1099/ijsem.0.002887
Hördt A, López MG, Meier-Kolthoff JP, Schleuning M, Weinhold L-M, Tindall BJ et al (2020) Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front Microbiol. https://doi.org/10.3389/fmicb.2020.00468
Kim Y-G, Hwang CY, Cho BC (2008) Pelagicola litoralis gen. nov., sp. nov., isolated from coastal water in Korea. Int J Syst Evol Microbiol 58:2102–2104. https://doi.org/10.1099/ijs.0.65820-0
Klotz F, Brinkhoff T, Freese HM, Wietz M, Teske A, Simon M, Giebel H-A (2018) Tritonibacter horizontis gen. nov., sp. nov., a member of the Rhodobacteraceae, isolated from the Deepwater Horizon oil spill. Int J Syst Evol Microbiol 68:736–744. https://doi.org/10.1099/ijsem.0.002573
Komagata K, Suzuki K-I (1988) Lipid and cell-wall analysis in bacterial systematics. Methods Microbiol 19:161–207. https://doi.org/10.1016/S0580-9517(08)70410-0
Kuypers MMM, Marchant HK, Kartal B (2018) The microbial nitrogen-cycling network. Nat Rev Microbiol 16:263–276. https://doi.org/10.1038/nrmicro.2018.9
Lefort V, Longueville JE, Gascuel O (2017) SMS: smart model selection in PhyML. Mol Biol Evol 34:2422–2424. https://doi.org/10.1093/molbev/msx149
Li Y, Xue H, Sang SQ, Lin CL, Wang XZ (2017) Phylogenetic analysis of family Neisseriaceae based on genome sequences and description of Populibacter corticis gen. nov., sp. nov., a member of the family Neisseriaceae, isolated from symptomatic bark of Populus x euramericana canker. PloS one 12:0174506. https://doi.org/10.1371/journal.pone.0174506
Lin CY, Zhang XY, Liu A, Liu C, Song XY, Su HN et al (2015) Marivirga atlantica sp. nov., isolated from seawater and emended description of the genus Marivirga. Int J Syst Evol Microbiol 65:1515–1519. https://doi.org/10.1099/ijs.0.000126
Luo H, Moran MA (2014) Evolutionary ecology of the marine Roseobacter clade. Microbiol Mol Biol Rev 78:573–587. https://doi.org/10.1128/mmbr.00020-14
Murray R, Doetsch R, Robinow C (1994) Determinative and cytolocal microscopy. In: Smibert RM, Krieg NR (eds) Methods for General and Molecular Bacteriology. Wiley, Washington, pp 607–654
Na SI, Kim YO, Yoon SH, Ha SM, Baek I, Chun J (2018) UBCG: Up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J Microbiol 56:280–285. https://doi.org/10.1007/s12275-018-8014-6
Ozeki S, Baba I, Takaya N, Shoun H (2001) A novel C1-using denitrifier Alcaligenes sp. STC1 and its genes for copper-containing nitrite reductase and azurin. Biosci Biotechnol Biochem 65:1206–1210. https://doi.org/10.1271/bbb.65.1206
Park S, Choi J, Won SM, Park JM, Yoon JH (2018) Aestuariibius insulae gen. nov., sp. nov., isolated from a tidal flat sediment. Int J Syst Evol Microbiol 68:1350–1355. https://doi.org/10.1099/ijsem.0.002679
Park S, Park JM, Choi SJ, Choi J, Yoon JH (2018) Pseudomaribius aestuariivivens gen. nov., sp. nov., isolated from a tidal flat sediment. Int J Syst Evol Microbiol 68:1344–1349. https://doi.org/10.1099/ijsem.0.002677
Park S, Yoon J-H (2013) Roseovarius sediminilitoris sp. nov., isolated from seashore sediment. Int J Syst Evol Microbiol 63:1741–1745. https://doi.org/10.1099/ijs.0.043737-0
Patureau D, Bernet N, Delgenes JP, Moletta R (2000) Effect of dissolved oxygen and carbon-nitrogen loads on denitrification by an aerobic consortium. Appl Microbiol Biotechnol 54:535–542. https://doi.org/10.1007/s002530000386
Qin QL, Xie BB, Zhang XY, Chen XL, Zhou BC, Zhou J et al (2014) A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol 196:2210–2215. https://doi.org/10.1128/jb.01688-14
Rodriguez-R LM, Konstantinidis KT (2016) The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. Peer J Preprints 4:e1900v1. https://doi.org/10.7287/peerj.preprints.1900v1
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. https://doi.org/10.1093/oxfordjournals.molbev.a040454
Sun Y, Li A, Zhang X, Ma F (2015) Regulation of dissolved oxygen from accumulated nitrite during the heterotrophic nitrification and aerobic denitrification of Pseudomonas stutzeri T13. Appl Microbiol Biotechnol 99:3243–3248. https://doi.org/10.1007/s00253-014-6221-6
Sun F, Wang B, Liu X, Lai Q, Du Y, Li G et al (2010) Leisingera nanhaiensis sp. nov., isolated from marine sediment. Int J Syst Evol Microbiol 60:275–280. https://doi.org/10.1099/ijs.0.010439-0
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. https://doi.org/10.1093/molbev/mst197
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L et al (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44:6614–6624. https://doi.org/10.1093/nar/gkw569
Tindall BJ, Sikorski J, Smibert RA, Krieg NR (2007) Phenotypic characterization and the principles of comparative systematics. In: Reddy CA (ed) Methods for General and Molecular Microbiology, 3rd edn. Wiley, Washington
Wang KL, Song ZM, Rong CH, Hao LY, Lai QL, Li SF, Xu Y (2018) Kandeliimicrobium roseum gen. nov., sp. nov., a new member of the family Rhodobacteraceae isolated from mangrove rhizosphere soil. Int J Syst Evol Microbiol 68:2158–2164. https://doi.org/10.1099/ijsem.0.002773
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703. https://doi.org/10.1128/jb.173.2.697-703.1991
Wirth JS, Whitman WB (2018) Phylogenomic analyses of a clade within the roseobacter group suggest taxonomic reassignments of species of the genera Aestuariivita, Citreicella, Loktanella, Nautella, Pelagibaca, Ruegeria, Thalassobius, Thiobacimonas and Tropicibacter, and the proposal of six novel genera. Int J Syst Evol Microbiol 68:2393–2411. https://doi.org/10.1099/ijsem.0.002833
Wu YH, Xu L, Zhou P, Wang CS, Oren A et al (2015) Brevirhabdus pacifica gen. nov., sp. nov., isolated from deep-sea sediment in a hydrothermal vent field. Int J Syst Evol Microbiol 64:3645–3651. https://doi.org/10.1099/ijsem.0.000469
Yoon S-H, Ha S-M, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617. https://doi.org/10.1099/ijsem.0.001755
Yu Z, Cao Y, Zhou G, Yin J, Qiu J (2018) Mangrovicoccus ximenensis gen. nov., sp. nov., isolated from mangrove forest sediment. Int J Syst Evol Microbiol 68:2172–2177. https://doi.org/10.1099/ijsem.0.002796
Zhang YJ, Liu XF, Kuang BZ, Zhang XY, Zhou MY, Chen S (2018) Neptunicoccus sediminis gen. nov., sp. nov., a member of the family Rhodobacteraceae isolated from the Yellow Sea. Int J Syst Evol Microbiol 68:1702–1706. https://doi.org/10.1099/ijsem.0.002728
Zhang YJ, Zhao JR, Zhang XY, Chen GZ, Zhou MY, Mo XH et al (2017) Euzebyella marina sp. nov., isolated from seawater. Int J Syst Evol Microbiol 67:920–924. https://doi.org/10.1099/ijsem.0.001712
Acknowledgements
The research was supported by the National Natural Science Foundation of China (31300005 and 81801982), the Bohai Sea and Yellow Sea Oceanological Comprehensive Scientific Investigation organized by the National Nature Science Foundation of China (41349901) and the Shandong Province Key Research and Development Program (Public welfare) (2017NC210001). The authors are grateful to Zhijuan Sun, Rongrong Ji, Bo Wang and Yuwei Du (School of Life Sciences, Qingdao Agricultural University, China) for experimental assistance. We also are grateful to Dr Xiaoyan He (Shandong University) for the analyses of respiratory quinone.
Author information
Authors and Affiliations
Contributions
Authors Ang Liu and Yan-Jiao Zhang designed and wrote the paper. Lili Xu and Yan-Jiao Zhang performed most of the laboratory experiments. Ang Liu analyzed the data. All authors contributed to manuscript revision and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xu, L., Liu, A. & Zhang, YJ. Zongyanglinia huanghaiensis gen. nov., sp. nov., a novel denitrifying bacterium isolated from the yellow sea, and transfer of Pelagicola marinus to Zongyanglinia gen. nov. as Zongyanglinia marinus comb. nov. Antonie van Leeuwenhoek 114, 137–149 (2021). https://doi.org/10.1007/s10482-020-01507-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-020-01507-1