
Abstract
Access to axenic cultures of Planctomycetes is crucial for further investigating their complex lifestyle, uncommon cell biology and primary and secondary metabolism. As a contribution to achieve this goal in the future, we here describe three strains belonging to the novel genus Novipirellula gen. nov. The strains were isolated from biotic and abiotic surfaces in the Baltic Sea and from the island Heligoland in the North Sea. Colony colours range from white to light pink. Cells are acorn-shaped and grew optimally at neutral pH and temperatures between 27 and 30 °C. Phylogenetic analyses revealed that the isolated strains represent three novel species belonging to a new genus, Novipirellula gen. nov. Beyond that, our analysis suggests that Rhodopirellula rosea LHWP3T, Rhodopirellula caenicola YM26-125T and Rhodopirellula maiorica SM1 are also members of this novel genus. Splitting the current genus Rhodopirellula into a more strictly defined genus Rhodopirellula and Novipirellula also allowed readjusting the genus threshold value for the gene rpoB, encoding the RNA polymerase β-subunit, which is used as phylogenetic marker for Planctomycetales. A threshold range of 75.5–78% identity of the analysed partial rpoB sequence turned out to be reliable for differentiation of genera within the family Planctomycetaceae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Planctomycetes are Gram-negative bacteria belonging to the PVC superphylum (Spring et al. 2016; Wagner and Horn 2006). In addition to Planctomycetes, the environmentally, medically and biotechnologically relevant PVC superphylum consists of the phyla Verrucomicrobia, Lentisphaerae, Kiritimatiellaeota, Candidatus Omnitrophica and Chlamydiae. Planctomycetes are ubiquitous microorganisms often found in aquatic environments, in which they play major roles in global carbon and nitrogen cycles. One important example are Planctomycetes capable of performing anaerobic ammonia oxidation (anammox) (Strous et al. 1999). By converting large amounts of ammonium to dinitrogen gas, anammox Planctomycetes find industrial applications for N-elimination during wastewater treatment (Peeters and van Niftrik 2018).
Planctomycetes share some characteristic morphological traits (Fuerst and Sagulenko 2011; König et al. 1984; Lonhienne et al. 2010) and were initially believed to link bacteria and eukaryotes (Fuerst and Sagulenko 2011). This picture changed with the advent of novel (super resolution) microscopic techniques and detailed physiological analyses of Planctomycetes (Jeske et al. 2015; Jogler et al. 2011; Jogler and Jogler 2013; Rivas-Marin et al. 2016; Van Teeseling et al. 2015), based on which the cell envelope architecture of Planctomycetes was classified as Gram-negative (Boedeker et al. 2017; Devos 2014). Nevertheless, Planctomycetes remain exceptional. For example, they divide by budding, binary fission or even a combination of both and lack proteins of the canonical divisome (such as FtsZ) (Wiegand et al. 2019). It was also found that many strains are resistant to several antibiotics (Cayrou et al. 2010; Godinho et al. 2019), either because of their degradation, intrinsic resistance or lack of targets (Jogler et al. 2012; Pilhofer et al. 2008).
Planctomycetes are not only found in ‘nutrient-rich’ suspensions such as wastewater, but also dwell in oligotrophic environments, e.g. seawater (Bengtsson et al. 2012; Bondoso et al. 2015; Bondoso et al. 2014; Bondoso et al. 2017; Lage and Bondoso 2014; Vollmers et al. 2017). Since decades, planctomycetal research is driven by the question how those microorganisms manage to survive in such rather nutrient-poor environments. To enable biomass formation and propagation, several Planctomycetes, e.g. strains of the Pirellula clade, attach to biotic surfaces such as algae or kelp and consume dissolved organic carbon released from these surfaces (Bondoso et al. 2015; Wiegand et al. 2019). The first strain of the Pirellula clade was isolated in 1973 and named ‘Pasteuria ramosa’ (Staley, 1973). Later, it was renamed first to ‘Pirella staleyi’ and finally to the currently valid name Pirellula staleyi (Schlesner and Hirsch, 1984; Schlesner and Hirsch, 1987). Twelve years later, a second species, ‘Pirella marina’ (now designated Blastopirellula marina), was isolated from brackish water of the Baltic Sea (Schlesner 1986). In a systematic analysis published in 2004, ninety-seven isolated budding strains and the above-mentioned species were classified in the already existing genus Pirellula and the novel genera Rhodopirellula and Blastopirellula based on DNA–DNA hybridization, physiological properties and chemotaxonomic tests (Schlesner et al. 2004). Morphological analyses took the mode of budding, bud shape, presence of fimbriae, crateriform structures and holdfast structure into account.
The overall analysis yielded important properties allowing to differentiate the type species of the three genera and comprise strain pigmentation (Rhodopirellula baltica is pink to red, Pirellula staleyi and Blastopirellula marina lack pigmentation), salt tolerance (R. baltica and B. marina tolerate high salinity conditions, P. staleyi shows only limited tolerance), substrate utilisation pattern and fatty acid composition (Schlesner et al. 2004). Another relevant criterion for differentiation is the relation to oxygen. Known Rhodopirellula and Blastopirellula species are strict aerobes, whereas some Pirellula species are also capable to grow under anoxic conditions (Ward et al. 2006).
In this study, we characterise the three strains Poly41T, Q31bT and Pla52oT which were isolated from natural or artificial surfaces in the North Sea and the Baltic Sea and analysed their phylogeny as well as basic phenotypic and genotypic characteristics. Based on the performed classification and by including information on already described species we were able to redefine the proposed genus threshold of the phylogenetic marker rpoB for the family Planctomycetaceae.
Materials and methods
Isolation of the strains
Strain Q31bT was sampled on the 5th of June 2013 from jellyfish (Aurelia aurita) on the north-western shore of Heligoland island in the North Sea (54.188 N, 7.875 E). Strain Poly41T was isolated from polystyrene particles embedded in an incubator in the water of the Baltic Sea for 14 days in 2 m depth at Heiligendamm Pier, Germany (54.146 N, 11.843 E). Collection date was the 8th of October 2015, when the incubator was removed from the water. Strain Pla52oT was isolated as described for strain Poly41T, but pieces of wood were embedded in the incubator instead of polystyrene particles. The incubation took place at exactly the same location (54.146 N, 11.843 E), but 1 year earlier (sampling date: 4. September 2014). Strains were isolated from the artificial (polystyrene) or natural (wood, jellyfish) sampling material as described in detail before (Wiegand et al. 2019). Initial amplification and sequencing of the 16S rRNA gene was performed as previously described (Rast et al. 2017). For subsequent cultivations M1 medium with HEPES as buffering agent and additionally supplemented with N-acetyl glucosamine (NAG) and artificial seawater (ASW) was used (designated M1H NAG ASW). Medium preparation was described previously (Kallscheuer et al. 2019a).
Light microscopy and scanning electron microscopy
Phase contrast microscopy and field emission scanning electron microscopy were performed according to protocols published earlier (Kallscheuer et al. 2019a).
Genome analysis
Genome information of the three isolated strains is available from GenBank under accession numbers SJPV00000000 (Poly41T), SJPY00000000 (Q31bT) and SJPT00000000 (Pla52oT). The corresponding 16S rRNA gene sequences are also available from GenBank under accession numbers MK554551 (Poly41T), MK554555 (Q31bT) and MK554549 (Pla52oT). Completeness and contamination of the genomes was determined using CheckM v1.0.131 (Parks et al. 2015). The primary metabolism was analysed by examining locally computed InterProScan (Mitchell et al. 2019) results cross-referenced with information from the UniProt database and BLASTp results of ‘typical’ protein sequences. The carbohydrate active enzymes were determined by employing dbCAN2 (Zhang et al. 2018), which automatically mines the CAZy database (Lombard et al. 2014).
Physiological analysis
For determination of the pH optimum 100 mM HEPES was used for cultivations at pH 7.0, 7.5 and 8.0. For cultivations at pH 5.0 and 6.0 HEPES was replaced by 100 mM 2-(N-morpholino)ethanesulfonic acid (MES), whereas 100 mM N-cyclohexyl-2-aminoethane-sulfonic acid (CHES) served as a buffering agent at pH 9.0 and 10.0. Cultivations for determination of the pH optimum were performed at 28 °C in triplicates. For determination of the temperature optimum the strains were cultivated at temperatures ranging from 10 to 40 °C in standard M1H NAG ASW medium at pH 7.5 in triplicates.
Phylogenetic analysis
16S rRNA gene phylogeny was computed for strains Poly41T, Q31bT and Pla52oT, the type strains of all described planctomycetal species (assessed in August 2019) and all isolates recently published (Wiegand et al. 2019), including the strains described recently (Boersma et al. 2019; Kallscheuer et al. 2019a, b, c; Kohn et al. 2019; Peeters et al. 2019). The 16S rRNA gene sequences were aligned with SINA (Pruesse et al. 2012). The phylogenetic analysis was done with RAxML (Stamatakis 2014) employing a maximum likelihood approach with 1000 bootstraps, the nucleotide substitution model GTR, gamma distributed rate variation and estimation of proportion of invariable sites (GTRGAMMAI option). Three 16S rRNA genes of bacterial strains from the PVC superphylum were used as outgroup. The average nucleotide identity (ANI) was calculated using OrthoANI (Lee et al. 2016) and the average amino acid identity (AAI) was calculated using the aai.rb script of the enveomics collection (Rodriguez-R and Konstantinidis 2016). The percentage of conserved proteins (POCP) was calculated as described (Qin et al. 2014). The rpoB nucleotide sequences were taken from the above mentioned as well as other publicly available genome annotations and the sequence identities were determined as described (Bondoso et al. 2013). Upon extracting only those parts of the sequence that would have been sequenced with the described primer set the alignment and matrix calculation was done with Clustal Omega (Sievers et al. 2011). For the multi-locus sequence analysis (MLSA) the unique single-copy core genome of all analysed genomes was determined with proteinortho5 (Lechner et al. 2011) with the ‘selfblast’ option enabled. The protein sequences of the resulting orthologous groups were aligned using MUSCLE v.3.8.31 (Edgar 2004). After clipping, partially aligned C- and N-terminal regions and poorly aligned internal regions were filtered using Gblocks (Castresana 2000). The final alignment was concatenated and clustered using the maximum likelihood method implemented by RAxML (Stamatakis 2014) with the ‘rapid bootstrap’ method and 500 bootstrap replicates.
Results and discussion
Phylogenetic analysis
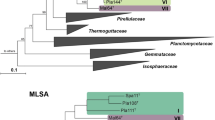
The phylogenetic positions of the three strains Poly41T, Q31bT and Pla52oT within the planctomycetal phylum were determined by 16S rRNA gene and MLSA analysis (Fig. 1). In the phylogenetic trees, the strains form a monophyletic cluster together with Rhodopirellula rosea LHWP3T, Rhodopirellula maiorica SM1 and Rhodopirellula caenicola YM26-125T (Richter et al. 2014; Roh et al. 2013; Yoon et al. 2014). Similarity of the 16S rRNA gene (Fig. 2, Table S1) shows that all strains of this cluster yield identity values of 94.5% or above, indicating that all strains belong to the same genus (Yarza et al. 2014). However, when including other described Rhodopirellula species (Rhodopirellula baltica, Rhodopirellula lusitana, Rhodopirellula europaea, Rhodopirellula islandica, Rhodopirellula bahusiensis, Rhodopirellula sallentina and Rhodopirellula rubra) into the analysis, minimal 16S rRNA gene identity values of 92.9% are observed. These values below the genus threshold are not only found when the novel strains described in this study are included, but also when analysing already published species. For example, R. lusitana and R. rosea share a 16S rRNA gene identity of 93.0%, indicating a relation on family-level rather than on genus-level. These findings suggest that the genus Rhodopirellula was defined too broadly.

MLSA- and 16S rRNA gene-based phylogeny. The phylogenetic trees show the position of the three here described strains in relation to their closest described relatives. Analysis was performed as described in the “Materials and methods” section. 16S rRNA gene phylogeny and MLSA were computed using the maximum likelihood method. Bootstrap values after 1000 re-samplings (16S rRNA gene)/500 re-samplings (MLSA) are given at the nodes (in %). The outgroup consists of three 16S rRNA genes from the PVC superphylum. In case of the MLSA tree ‘Bythopirellula goksoyri’ Pr1d served as outgroup

Phylogeny of the novel genus comprising strains Q31bT, Poly41T and Pla52oT as well as the former Rhodopirellula strains R. rosea LHWP3T and R. caenicola YM26-125T. The black font gives the 16S rRNA gene identity and the orange font gives the whole genome-based average nucleotide identity (ANI) values. Comparison of rpoB similarity values could not be performed for R. rosea LHWP3T and R. caenicola YM26-125T as their genome sequences are not available
Based on the 16S rRNA gene identity values, we propose that the novel strains Poly41T, Q31bT and Pla52oT form the novel genus Novipirellula gen. nov. and that R. rosea LHWP3T, R. maiorica SM1 and R. caenicola YM26-125T should be transferred to this novel genus. Supported by a minimal 16S rRNA gene identity of 95.4%, R. baltica, R. lusitana, R. europaea, R. islandica, R. bahusiensis, R. sallentina and R. rubra should remain in the now more strictly defined Rhodopirellula genus.
When comparing two-way AAI values for all strains described as Rhodopirellula and the novel isolates (Table S2), the minimal obtained value is 50.5%. The same analysis for the newly defined Rhodopirellula and Novipirellula genera gives minimal AAI values of 57.8% and 58.4%, respectively. Although all of these values are below the proposed AAI genus threshold of 60–80%, only the latter are still well within the ranges found for different bacterial genera (Luo et al. 2014). A similar pattern is found for POCP (Table S3). The larger clade comprising all strains has a minimal POCP of 44.1%, a value distinctly below the proposed genus threshold of 50% (Qin et al. 2014). The now smaller Rhodopirellula taxon and the Novipirellula reach higher minimal POCP values of 54.1% and 47.5%, respectively, which are either above or at least closer to the proposed border. In brief, the separation of the current genus Rhodopirellula to a more strictly defined genus Rhodopirellula and the novel genus Novipirellula is supported on genomic level.
While strains R. rosea LHWP3T, R. caenicola YM26-125T and R. maiorica SM1 are members of the novel genus, the latter is not a validly described species and is therefore excluded from more detailed taxonomic analyses. For the two former and our three isolates Poly41T, Q31bT and Pla52oT, the identities of the 16S rRNA gene sequences range between 94.5% and 98.1% (Fig. 2). All values are below the species threshold of 98.7% (Stackebrandt and Ebers 2006), indicating that all five strains, including the novel isolates, represent individual species belonging to the same genus. For the novel strains with sequenced genomes, this finding is supported by ANI values (Fig. 2) unambiguously below the species threshold of 95% (Kim et al. 2014).
New thresholds for rpoB as marker gene
Previously, the comparison of a partial sequence of the rpoB gene encoding the β-subunit of RNA polymerase was proposed as reliable molecular marker to infer phylogeny of species of the order Planctomycetales (Bondoso et al. 2013). A threshold value of 72% identity was proposed for differentiating between genera of this order. To a certain extent, this analysis was built on strains of the now split genus Rhodopirellula. To adapt the threshold values to the reclassified taxa as well as to include all strains belonging to the family Planctomycetaceae recently published (Wiegand et al. 2019), we re-evaluated the partial rpoB sequence used earlier (Bondoso et al. 2013) (Table S4). Based on this analysis, we here propose a new genus threshold range of 75.5–78.0% partial rpoB sequence identity. The novel genus Novipirellula and the re-defined genus Rhodopirellula are also supported by this threshold. The rpoB-based species threshold found has not been affected by our analysis (Bondoso et al. 2013).
Morphological and physiological analysis
We employed both light microscopy (Fig. 3) and scanning electron microscopy (Fig. 4) to analyse the morphologies of Poly41T, Q31bT and Pla52oT cells during the exponential growth phase. Detailed information on morphology and cell division is summarised in Table 1 and compared to closely related strains.

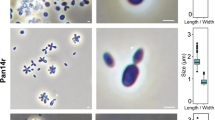
Light microscopy images and cell size plots of strains Poly41T, Q31bT and Pla52oT. The figure highlights the mode of cell division (a, c, e) and gives a general overview of cell morphology (b, d, f). The scale bar is 1 µm. Cell sizes (g, h, i) of at least 100 representative cells were counted manually or by using a semi-automated object count tool

Scanning electron microscopic pictures of strains Poly41T, Q31bT and Pla52oT. The scale bar is 1 µm
Cells of strain Poly41T have a size of 2.0 ± 0.4 × 1.0 ± 0.2 µm (Fig. 3c) and show a characteristic acorn-shape with a polar ‘fibre cap’ covering up to 40% of the cell surface. Cells either live planktonic or form small rosettes of 3–8 cells (Fig. 4b). Division occurs by polar budding with the daughter cell having a round shape. Crateriform structures and flagella were observed. Colonies have a light pink colour. The preferred temperature and pH are 27 °C and 7.5, respectively, while growth in a range of 15–27 °C and of pH 6.5–8.5 was observed (Fig. 5). Growth is thus mesophilic and neutrophilic. A maximal growth rate of 0.039 h1 (generation time of 18 h) was observed during cultivation.

Temperature and pH profiles of strains Poly41T, Q31bT and Pla52oT. In the upper panel, the given data points show the average growth rates obtained after cultivation of the three isolated strains in M1H NAG ASW medium in biological triplicates at different temperatures and a constant pH of 7.5. In the bottom panel, the data points show the average growth rates for cultivation at different pH values at a constant temperature of 28 °C
Q31bT has a similar cell shape as Poly41T but the cells are slightly smaller (1.6 ± 0.3 × 0.8 ± 0.1 µm) (Fig. 3f). Aggregate formation is more pronounced for Q31bT as aggregates consisting of 10 to more than 50 individual cells were found (Fig. 4d, e). Mode of division and shape of the daughter cell are comparable to Poly41T. The temperature optimum at 30 °C and the pH optimum at pH 7.5 are also similar to Poly41T, but growth of strain Q31bT was observed in a distinctly broader temperature (10–36 °C) and pH range (pH 5.5–9.0). The maximal growth rate of 0.018 h−1 (generation time of 39 h) was about half of the maximal growth rate of Poly41T (Fig. 5).
Cells of strain Pla52oT have the same colour, size and mode of division as Poly41T. The cell shape also resembles an acorn, but cells appear elongated and more strongly deformed during SEM analysis compared to Poly41T and Q31bT (Fig. 4g). The number of cells per aggregate (typically 10–30) was more uniform compared to the other two strains. Pla52oT showed the same temperature and pH profile as Q31bT and, with µmax = 0.078 h−1 (generation time of 9 h), the fastest growth of the three isolates (Fig. 5).
With regard to shape and temperature/pH preferences there are only slight differences when comparing the isolated strains with the related species ‘Novipirellula maiorica’ SM1, ‘Novipirellula rosea’ LHWP3T and ‘Novipirellula caenicola’ YM26-125T (Table 1). The already published strains are slightly smaller and have more roundish shape than the strains described in this study. The degree of pigmentation within the genus Novipirellula varies from white to red.
Genomic characteristics
The genomic characteristics of the three isolated strains are summarised in Table 1. As ‘N. rosea’ LHWP3T and ‘N. caenicola’ YM26-125T have not been sequenced yet, obtained genome data could only be compared to ‘N. maiorica’ SM1. All strains have a similar G + C content of 53–58%, but differ significantly in their genome sizes. While Q31bT and Pla52oT have a genome size of 7.3–7.4 Mb, about 20–25% larger genomes are present in Poly41T and ‘N. maiorica’ SM1 (9.2 and 8.9 Mb, respectively). These differences in genome size are not reflected in the strain relationships as Poly41T clustered with Q31bT in our phylogenetic analysis and both are more distantly related to Pla52oT. Larger genomes correlated with higher numbers of open reading frames considering that all strains had similar coding densities (86–89%) and protein-coding genes per Mb (770–823). The number of tRNA genes is identical for Poly41T and Q31bT (78 tRNA genes), but is significantly lower in Pla52oT (51 tRNA genes) and higher in ‘N. maiorica SM1’ (95 tRNA genes). All sequenced strains harbour one copy of the 16S rRNA gene.
Genome-based analysis of the central carbon metabolism
Based on the genome sequences of the three novel isolates and of ‘N. maiorica SM1’ for comparison we investigated the presence of pathways representing the microbial central carbon metabolism (Table 2). A complete set of genes coding for glycolytic enzymes of the common Embden–Meyerhof–Parnas pathway is present in all strains, while also hits for key enzymes of the Entner–Doudoroff pathway were found. Additionally, degradation of sugars via the pentose phosphate pathway appears to be possible in all four strains. This was somehow expected as important precursors for the biosynthesis of amino acids and nucleotides branch off from this pathway and an incomplete or inactive pentose phosphate pathway would be associated to multiple auxotrophies. Our analysis further suggests that all strains harbour a complete TCA cycle, except for Pla52oT, in which we could not identify the gene encoding isocitrate dehydrogenase. This, however, might be explained by the non-closed genomes with several scaffolds. In contrast to many other bacteria, Novipirellula strains seem to lack the glyoxylate shunt, which is typically utilised as anaplerotic pathway during growth on acetate or for enabling biomass formation from acetyl-CoA obtained e.g. during β-oxidation of fatty acids. Novipirellula species might utilise alternative pathways for biomass formation from acetate or are unable to use acetate or fatty acids as sole carbon and energy source. The capability of de novo glucose biosynthesis should be given as genes required for a functional gluconeogenesis pathway are present.
Monomeric sugars as easily degradable carbon sources are typically not found in larger amounts in oligotrophic environments which Planctomycetes inhabit. For enabling breakdown of complex polysaccharides obtained from biotic surfaces, Planctomycetes harbour larger sets of glycolytic enzymes, which convert complex and decorated polysaccharides to monomeric units, which can then be channelled into the central carbon metabolism. For getting a first insight into the enzymatic set of Novipirellula strains, we analysed the most common classes of such glycolytic enzymes and compared the numbers of predicted enzymes in the four strains (Table 3). In our analysis, Poly41T clearly stands out. With over 500 predicted glycolytic enzymes (of which 329 are putative glycoside hydrolases) it has by far the highest number compared to the other three strains, which have 230–330 putative glycolytic enzymes. These numbers also correlate with the genome size as Poly41T also has the largest genome (9.20 Mb) of the investigated strains. ‘N. maiorica’ SM1 (8.87 Mb) has the second largest genome and the total number of glycolytic enzymes is above 300, whereas it is below 300 for the other two strains. Taken together, a high number of glycolytic enzymes suggests large potential of Novipirellula strains for degradation of a broad range of different polymeric compounds with different sugar compositions and decorations of the sugar units.
Genes potentially involved in secondary metabolite production
For assessing the relevance of the three characterised strains as potential sources of novel secondary metabolites we performed AntiSMASH analyses based on the genome sequence of Poly41T, Q31bT and Pla52oT and N. maiorica SM1 (Blin et al. 2019). In most cases, secondary metabolites are produced by dedicated enzymes of the families of polyketide synthases (PKS) or non-ribosomal peptide synthetases (NRPS). PKSs use coenzyme A (CoA)-activated carboxylic acids as substrates and often malonyl-CoA for carbon chain elongation while NRPSs condense proteinogenic or non-proteinogenic amino acids. We cannot exclude that Planctomycetes follow additional, yet unknown, strategies for secondary metabolite production, however relevant proteins most likely escaped the AntiSMASH analysis. According to our analysis, all four strains harbour two genes involved in terpenoid production (Table 3). None of the strains encodes an NRPS, however two putative mixed type I PKS-NRPS proteins are encoded in Poly41T and Q31bT. One or two putative type I PKS genes were identified in all strains except Pla52oT, no type II PKSs are present, and a single putative type III PKS is encoded by all strains except ‘N. maiorica’ SM1 (Table 3). Clusters involved in the production of bacteriocins, resorcinol or ectoine were not identified in the four genomes. Q31bT and Poly41T encode the same set of identified clusters involved in secondary metabolite production, which differs from the sets encoded in Pla52oT and ‘N. maiorica’ SM1. This observation also reflects the close relationship of Poly41T and Q31bT compared to Pla52oT and ‘N. maiorica’ SM1, which is in line with the obtained results during the phylogenetic analysis.
Conclusion
Differentiation of the genera Pirellula, Rhodopirellula, Blastopirellula and Novipirellula
In addition to the genetic differentiation of the genera Pirellula, Rhodopirellula, Blastopirellula and Novipirellula based on different phylogenetic markers, attention should also be given to major differences in physiological features separating the respective type species of the genera from each other. Key physiological features are compared in Table 4. Major differences can be observed in cell size, strain pigmentation, relation to oxygen, salinity and polar lipid composition. Major fatty acids are very similar when comparing the type species of the four genera, but utilisation pattern of sugars could be used for strain differentiation, e.g. when investigating growth on D-mannitol/gluconate (N. rosea: +/−, R. baltica: −/+, B. marina: +/+, P. staleyi: −/−).
Based on our physiological and phylogenetic analysis of three novel isolates and of the transferred strains, we here introduce the novel genus Novipirellula gen. nov., which is separated from the genera Rhodopirellula, Blastopirellula and Pirellula by phylogenetic and physiological differences. We propose the names Novipirellula artificiosorum Poly41T, Novipirellula aureliae Q31bT and Novipirellula galeiformis Pla52oT for the novel isolates. Our analyses suggest that the strains Rhodopirellula maiorica SM1, Rhodopirellula rosea LHWP3T and Rhodopirellula caenicola YM26-125T also belong to the novel genus Novipirellula gen. nov. and are thus reclassified.
Description of Novipirellula gen. nov.
Novipirellula (No.vi.pi.rel’lu.la. L. masc. adj. novus new; N.L. fem. n. Pirellula name of a bacterial genus; N.L. fem. n. Novipirellula a new type of Pirellula). Species of this genus are Gram-negative, aerobic, mesophilic, neutrophilic and heterotrophic. Cells are ovoid or acorn-shaped, motile and divide by budding. Colony colours range from white to red. The G + C content is between 51 and 59%. We introduce Novipirellula rosea as type species of the genus as it was the first validly described species of this genus (Roh et al. 2013).
Description of Novipirellula artificiosorum sp. nov.
Novipirellula artificiosorum (ar.ti.fi.ci.o.so’rum. L. masc. adj. artificiosus artificial, not natural; N.L. gen. pl. n. artificiosorum of artificial, not natural things, referring to the isolation of the strain from plastic). Cells are acorn-shaped (length: 2.0 ± 0.4 µm, width: 1.0 ± 0.2 µm), form aggregates and divide by polar budding. Colonies have a light pink colour. The type strain grows at ranges of 15–27 °C (optimum 27 °C) and at pH 6.5–8.5 (optimum 7.5). The type strain genome has a size of 9,199,719 bp and a G + C content of 55.3 ± 1.6%. The genome (acc. no. SJPV00000000) and 16S rRNA gene sequences (acc. no. MK554551) of the type strain are available from GenBank.
The type strain is Poly41T (DSM 103145T = VKM B-3437T) isolated from polystyrene particles incubated in the Baltic Sea close to a landing stage in Heiligendamm, Germany.
Description of Novipirellula aureliae sp. nov.
Novipirellula aureliae (au.re’li.ae. N.L. gen. n. aureliae of Aurelia; corresponding to the isolation of the strain from the common jellyfish Aurelia aurita). Cells are acorn-shaped (length: 1.6 ± 0.3, width: 0.8 ± 0.1 µm), form aggregates and divide by polar budding. Lucid white colonies are formed. The temperature optimum of the type strain is 30 °C (growth observed from 10–36 °C). The preferred pH is 7.5, but growth is also observed at pH 5.5–9.0. The genome of the type strain has a size of 7,267,653 bp and a G + C content of 52.9 ± 2.2%. The type strain genome (acc. no. SJPY00000000) and 16S rRNA gene sequence (acc. no. MK554555) are available from the GenBank database.
The type strain is Q31bT (DSM 103929T = LMG 29701T) isolated from jellyfish Aurelia aurita on the shore of the island Heligoland.
Description of Novipirellula galeiformis sp. nov.
Novipirellula galeiformis (ga.le.i.for’mis. L. fem. n. galea a helmet; L. suff. adj. formis a form, a figure; N.L. fem. adj. galeiformis shaped like a helmet; corresponding to the helmet-shaped formation of the fibres). Cells are acorn-shaped (1.9 ± 0.4 µm x 1.0 ± 0.2 µm), form aggregates and divide by polar budding. Colonies have a light pink colour. The preferred temperature and pH of the type strain are 30 °C and 7.5, respectively, while growth is observed in the range of 10–36 °C and at pH 6.0–9.0. The type strain genome has a size of 7,403,604 bp and a G + C content of 55.8 ± 2.2%. The genome (acc. no. SJPT00000000) and 16S rRNA gene sequence (acc. no. MK554549) are available from GenBank.
The type strain is Pla52oT (DSM 103357T = LMG 29744T; synonym Pla52Original) isolated from wood incubated in the Baltic Sea in 2 m depth near Heiligendamm, Germany.
Description of Novipirellula rosea comb. nov.
Basonym: Rhodopirellula rosea Roh et al. 2014
Strain characteristics as described before (Roh et al. 2013). The type strain is LHWP3T (KACC 15560T = JCM 17759T).
Description of Novipirellula caenicola comb. nov.
Basonym: Rhodopirellula caenicola Yoon et al. 2015
Strain characteristics as described before (Yoon et al. 2014). The type strain is YM26-125T (KCTC 32995T = NBRC 110016T).
References
Bengtsson MM, Sjøtun K, Lanzén A, Øvreås L (2012) Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea. ISME J 6:2188–2198
Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY, Medema MH, Weber T (2019) antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucl Acids Res 47:W81–W87
Boedeker C, Schuler M, Reintjes G, Jeske O, van Teeseling MC, Jogler M, Rast P, Borchert D, Devos DP, Kucklick M, Schaffer M, Kolter R, van Niftrik L, Engelmann S, Amann R, Rohde M, Engelhardt H, Jogler C (2017) Determining the bacterial cell biology of Planctomycetes. Nat Commun 8:14853
Boersma A, Kallscheuer N, Wiegand S, Rast R, Peeters S, Mesman R, Heuer A, Boedeker C, Jetten M, Rohde M, Jogler M, Jogler C (2019) Alienimonas californiensis gen. nov. sp. nov., a novel Planctomycete isolated from the kelp forest in Monterey Bay. Antonie van Leeuwenhoek, https://doi.org/10.1007/s10482-019-01367-4
Bondoso J, Harder J, Lage OM (2013) rpoB gene as a novel molecular marker to infer phylogeny in Planctomycetales. Antonie Van Leeuwenhoek 104:477–488
Bondoso J, Balague V, Gasol JM, Lage OM (2014) Community composition of the Planctomycetes associated with different macroalgae. FEMS Microbiol Ecol 88:445–456
Bondoso J, Albuquerque L, Nobre MF, Lobo-da-Cunha A, da Costa MS, Lage OM (2015) Roseimaritima ulvae gen. nov., sp. nov. and Rubripirellula obstinata gen. nov., sp. nov. two novel planctomycetes isolated from the epiphytic community of macroalgae. Syst Appl Microbiol 38:8–15
Bondoso J, Godoy-Vitorino F, Balague V, Gasol JM, Harder J, Lage OM (2017) Epiphytic Planctomycetes communities associated with three main groups of macroalgae. FEMS Microbiol Ecol 93: fiw255
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Cayrou C, Raoult D, Drancourt M (2010) Broad-spectrum antibiotic resistance of Planctomycetes organisms determined by Etest. J Antimicrob Chemother 65:2119–2122
Devos DP (2014) Re-interpretation of the evidence for the PVC cell plan supports a Gram-negative origin. Antonie Van Leeuwenhoek 105:271–274
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32:1792–1797
Frank CS (2011) Polyphasische Taxonomie, Kerngenom und Lebenszyklus von Rhodopirellula-Stämmen. Ph. D. thesis, University of Bremen, Germany
Fuerst JA, Sagulenko E (2011) Beyond the bacterium: Planctomycetes challenge our concepts of microbial structure and function. Nat Rev Microbiol 9:403–413
Godinho O, Calisto R, Ovreas L, Quinteira S, Lage OM (2019) Antibiotic susceptibility of marine Planctomycetes. Antonie Van Leeuwenhoek 112:1273–1280
Jeske O, Schüler M, Schumann P, Schneider A, Boedeker C, Jogler M, Bollschweiler D, Rohde M, Mayer C, Engelhardt H (2015) Planctomycetes do possess a peptidoglycan cell wall. Nat Commun 6:7116
Jogler M, Jogler C (2013) Towards the development of genetic tools for Planctomycetes. In: Fuerst JA (ed) Planctomycetes: cell structure, origins and biology. Springer, New York, pp 141–164
Jogler C, Glöckner FO, Kolter R (2011) Characterization of Planctomyces limnophilus and development of genetic tools for its manipulation establish it as a model species for the phylum Planctomycetes. Appl Environ Microbiol 77:5826–5829
Jogler C, Waldmann J, Huang X, Jogler M, Glöckner FO, Mascher T, Kolter R (2012) Identification of proteins likely to be involved in morphogenesis, cell division, and signal transduction in Planctomycetes by comparative genomics. J Bacteriol 194:6419–6430
Kallscheuer N, Jogler M, Wiegand S, Peeters S, Heuer A, Boedeker C, Jetten M, Rohde M, Jogler C (2019a) Rubinisphaera italica sp. nov. isolated from a hydrothermal area in the Tyrrhenian Sea close to the volcanic island Panarea. Antonie van Leeuwenhoek. https://doi.org/10.1007/s10482-019-01329-w
Kallscheuer N, Jogler M, Wiegand S, Peeters S, Heuer A, Boedeker C, Jetten M, Rohde M, Jogler C (2019b) Three novel Rubripirellula species isolated from artificial plastic surfaces submerged in the German part of the Baltic Sea and the estuary of the river Warnow. Antonie van Leeuwenhoek, https://doi.org/10.1007/s10482-019-01368-3
Kallscheuer N, Wiegand S, Jogler M, Boedeker C, Peeters S, Rast P, Heuer A, Jetten M, Rohde MCJ (2019c) Rhodopirellula heiligendammensis sp. nov., Rhodopirellula pilleata sp. nov., and Rhodopirellula solitaria sp. nov. isolated from natural or artificial marine surfaces in Northern Germany and California, USA. Antonie van Leeuwenhoek, https://doi.org/10.1007/s10482-019-01366-5
Kim M, Oh HS, Park SC, Chun J (2014) Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol 64:346–351
Kohn T, Wiegand S, Boedeker C, Rast P, Heuer A, Jetten MSM, Schüler M, Becker S, Rohde C, Müller R-W, Brümmer F, Rohde M, Engelhardt H, Jogler M, Jogler C (2019) Planctopirus ephydatiae, a novel Planctomycete species isolated from the freshwater sponge Ephydatia fluviatilis. Syst Appl Microbiol. https://doi.org/10.1016/j.syapm.2019.126022
König E, Schlesner H, Hirsch P (1984) Cell wall studies on budding bacteria of the Planctomyces/Pasteuria group and on a Prosthecomicrobium sp. Arch Microbiol 138:200–205
Lage OM, Bondoso J (2014) Planctomycetes and macroalgae, a striking association. Front Microbiol 5:267
Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ (2011) Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinform 12:124
Lee I, Ouk Kim Y, Park SC, Chun J (2016) OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol 66:1100–1103
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucl Acids Res 42:D490–D495
Lonhienne TG, Sagulenko E, Webb RI, Lee K-C, Franke J, Devos DP, Nouwens A, Carroll BJ, Fuerst JA (2010) Endocytosis-like protein uptake in the bacterium Gemmata obscuriglobus. Proc Natl Acad Sci USA 107:12883–12888
Luo C, Rodriguez RL, Konstantinidis KT (2014) MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucl Acids Res 42:e73
Mitchell AL, Attwood TK, Babbitt PC et al (2019) InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucl Acids Res 47:D351–D360
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055
Peeters SH, van Niftrik L (2018) Trending topics and open questions in anaerobic ammonium oxidation. Curr Opin Chem Biol 49:45–52
Peeters S, Wiegand S, Kallscheuer N, Jogler M, Heuer A, Jetten M, Rast P, Boedeker C, Rohde M, Jogler C (2019) Three marine strains constitute the novel genus and species Crateriforma conspicua gen. nov. sp. nov. in the phylum Planctomycetes. Antonie van Leeuwenhoek, accepted manuscript (ANTO-D-19-00328)
Pilhofer M, Rappl K, Eckl C, Bauer AP, Ludwig W, Schleifer KH, Petroni G (2008) Characterization and evolution of cell division and cell wall synthesis genes in the bacterial phyla Verrucomicrobia, Lentisphaerae, Chlamydiae, and Planctomycetes and phylogenetic comparison with rRNA genes. J Bacteriol 190:3192–3202
Pruesse E, Peplies J, Glöckner FO (2012) SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829
Qin QL, Xie BB, Zhang XY, Chen XL, Zhou BC, Zhou J, Oren A, Zhang YZ (2014) A proposed genus boundary for the prokaryotes based on genomic insights. J Bacteriol 196:2210–2215
Rast P, Glöckner I, Boedeker C, Jeske O, Wiegand S, Reinhardt R, Schumann P, Rohde M, Spring S, Glöckner FO (2017) Three novel species with peptidoglycan cell walls form the new genus Lacunisphaera gen. nov. in the family Opitutaceae of the Verrucomicrobial subdivision 4. Front Microbiol 8:202
Richter M, Richter-Heitmann T, Klindworth A, Wegner C-E, Frank CS, Harder J, Glöckner FO (2014) Permanent draft genomes of the Rhodopirellula maiorica strain SM1. Mar Genomics 13:19–20
Rivas-Marin E, Canosa I, Santero E, Devos DP (2016) Development of genetic tools for the manipulation of the Planctomycetes. Front Microbiol 7:914
Rodriguez-R LM, Konstantinidis KT (2016) The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ (preprints)
Roh SW, Lee H-W, Yim KJ, Shin N-R, Lee J, Whon TW, Lim N-L, Kim D, Bae J-W (2013) Rhodopirellula rosea sp. nov., a novel bacterium isolated from an ark clam Scapharca broughtonii. J Microbiol 51:301–304
Schlesner H (1986) Pirella marina sp. nov., a budding, peptidoglycan-less bacterium from brackish water. Syst Appl Microbiol 8:177–180
Schlesner H, Hirsch P (1984) Assignment of ATCC 27377 to Pirella gen. nov. as Pirella staleyi comb. nov. Int J Syst Evol Microbiol 34:492–495
Schlesner H, Hirsch P (1987) Rejection of the genus name Pirella for pear-shaped budding bacteria and proposal to create the genus Pirellula gen. nov. Int J Syst Evol Microbiol 37:441
Schlesner H, Rensmann C, Tindall BJ, Gade D, Rabus R, Pfeiffer S, Hirsch P (2004) Taxonomic heterogeneity within the Planctomycetales as derived by DNA–DNA hybridization, description of Rhodopirellula baltica gen. nov., sp. nov., transfer of Pirellula marina to the genus Blastopirellula gen. nov. as Blastopirellula marina comb. nov. and emended description of the genus Pirellula. Int J Syst Evol Microbiol 54:1567–1580
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Spring S, Bunk B, Spröer C, Schumann P, Rohde M, Tindall BJ, Klenk H-P (2016) Characterization of the first cultured representative of Verrucomicrobia subdivision 5 indicates the proposal of a novel phylum. ISME J 10:2801
Stackebrandt E, Ebers J (2006) Taxonomic parameter revisited: tarnished gold standards. Microbiol Today 33:152–155
Staley JT (1973) Budding bacteria of the Pasteuria-Blastobacter group. Can J Microbiol 19:609–614
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Strous M, Fuerst JA, Kramer EH, Logemann S, Muyzer G, van de Pas-Schoonen KT, Webb R, Kuenen JG, Jetten MS (1999) Missing lithotroph identified as new planctomycete. Nature 400:446–449
Van Teeseling MC, Mesman RJ, Kuru E, Espaillat A, Cava F, Brun YV, Van Nieuwenhze MS, Kartal B, Van Niftrik L (2015) Anammox Planctomycetes have a peptidoglycan cell wall. Nat Commun 6:6878
Vollmers J, Frentrup M, Rast P, Jogler C, Kaster AK (2017) Untangling genomes of novel planctomycetal and Verrucomicrobial species from monterey bay kelp forest metagenomes by refined binning. Front Microbiol 8:472
Wagner M, Horn M (2006) The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opin Biotechnol 17:241–249
Ward N, Staley JT, Fuerst JA, Giovannoni S, Schlesner H, Stackebrandt E (2006) The order Planctomycetales, including the genera Planctomyces, Pirellula, Gemmata and Isosphaera and the Candidatus genera Brocadia, Kuenenia and Scalindua. Prokaryotes 7:757–793
Wiegand S, Jogler M, Boedeker C, Pinto D, Vollmers J, Rivas-Marín E, Kohn T, Peeters SH, Heuer A, Rast P, Oberbeckmann S, Bunk B, Jeske O, Meyerdierks A, Storesund JE, Kallscheuer N, Lücker S, Lage OM, Pohl T, Merkel BJ, Hornburger P, Müller R-W, Brümmer F, Labrenz M, Spormann AM, Op den Camp HJM, Overmann J, Amann R, Jetten MSM, Mascher T, Medema MH, Devos DP, Kaster A-K, Øvreås L, Rohde M, Galperin MY, Jogler C (2019) Cultivation and functional characterization of 79 planctomycetes uncovers their unique biology. Nat Microbiol. https://doi.org/10.1038/s41564-019-0588-1
Winkelmann N, Harder J (2009) An improved isolation method for attached-living Planctomycetes of the genus Rhodopirellula. J Microbiol Meth 77:276–284
Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, Whitman WB, Euzeby J, Amann R, Rossello-Mora R (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645
Yoon J, Matsuo Y, Kasai H, Lee M-K (2014) Phylogenetic and taxonomic analyses of Rhodopirellula caenicola sp. nov., a new marine Planctomycetes species isolated from Iron Sand. J Phylogen Evol Biol 3:143
Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y (2018) dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucl Acids Res 46:W95–W101
Acknowledgements
Part of this research was funded by the Deutsche Forschungsgemeinschaft Grants KA 4967/1-1 and JO 893/4-1, Grant ALWOP.308 of the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO), SIAM (Soehngen Institute for Anaerobic Microbiology) Grant no. 024002002 and the Radboud Excellence fellowship. We thank Ina Schleicher for skillful technical assistance. We also thank our collaborators Sonja Oberbeckmann, Matthias Labrenz (IOW Warnemünde, Germany), Jörn Petersen (DSMZ) and the Biological Institute Heligoland (BAH) for sampling support. We thank Brian Tindall and Regine Fähnrich as well as the BCCM/LMG Bacteria collection for on-going support during strain deposition.
Author information
Authors and Affiliations
Contributions
NK and SW wrote the manuscript, analysed data and prepared figures, AH, PR and MJ isolated the strains and performed the initial cultivation and strain deposition, SHP and CB performed the light microscopic analysis, MSMJ contributed to text preparation and revised the manuscript, MR performed the electron microscopic analysis, CJ, PR and AH took the samples in the Baltic Sea and on Heligoland, and CJ supervised the study. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
This article does not contain any studies with animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kallscheuer, N., Wiegand, S., Peeters, S.H. et al. Description of three bacterial strains belonging to the new genus Novipirellula gen. nov., reclassificiation of Rhodopirellula rosea and Rhodopirellula caenicola and readjustment of the genus threshold of the phylogenetic marker rpoB for Planctomycetaceae. Antonie van Leeuwenhoek 113, 1779–1795 (2020). https://doi.org/10.1007/s10482-019-01374-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-019-01374-5