
Abstract
Organized bacterial communities, or biofilms, provide an important reservoir for persistent cells that are inaccessible or tolerant to antibiotics. Curli pili are cell-surface structures produced by certain bacteria and have been implicated in biofilm formation in these species. In order to determine whether these structures, which were suggested to be encoded by the Rv3312A (mtp) gene, have a similar role in Mycobacterium tuberculosis, we generated a Δmtp mutant and a mtp-complemented strain of a clinical isolate of M. tuberculosis and analyzed these strains for their ability to produce pili in comparison to the wild-type strain. Phenotypic analysis by transmission electron microscopy proved the essentiality of mtp for piliation in M. tuberculosis. We then compared biofilm formation of the derived strains in detergent-free Sauton’s media. Biofilm mass was quantified spectrophotometrically using crystal violet. Furthermore, we examined mtp gene expression by quantitative real-time PCR in wild-type cells grown under biofilm versus planktonic growth conditions. We found a 68.4 % reduction in biofilm mass in the mutant compared to the wild-type strain (P = 0.002). Complementation of the mutant resulted in a restoration of the wild-type biofilm phenotype (P = 0.022). We, however, found no significant difference between mtp expression in cells of the biofilm to those growing planktonically. Our findings highlight a crucial, but non-specific, role of pili in the biofilm lifestyle of M. tuberculosis and indicate that they may represent an important target for the development of therapeutics to attenuate biofilm formation, thereby potentially reducing persistence.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tuberculosis (TB) has been, and continues to be, one of the most widespread bacterial infections world-wide. The standard treatment for TB patients involves the use of multiple antibiotics for 6 months and the regimen for most patients with drug-resistant TB takes 20 months (WHO 2012). Whilst this rigorous approach to treatment has resulted in cure for close to 36 million people, 1.8 million people still die of the disease each year (Sasindran and Torrelles 2011). This is largely due to the prolonged therapy that results in patient non-compliance and challenges in delivery of long-term treatment in socioeconomically- and medically-poor countries with a high burden of disease (Spigelman and Ma 2004). Improved TB control is dependent on the development of newer drugs that can clear infection efficiently and within a shorter duration. This in-turn is dependent on a greater understanding into the mechanisms employed by the causative organism, Mycobacterium tuberculosis, to persist against the challenges of the competent host immune system and antibiotic pressure (Ojha and Hatfull 2012).
The most accepted hypothesis for the extensive TB treatment regimen is the existence of persisters, a sub-group of microorganisms that are phenotypically resistant to, or tolerant of, the commonly used antibiotics (Spigelman and Ma 2004). Microbial species are known to form biofilms (O’Toole et al. 2000). These self-assembled, organized, multicellular, and matrix-encapsulated structures are contributors to microbial persistence, promoting survival of its inhabitants in the face of environmental stresses (Costerton et al. 1999). Ojha et al. (2008) provided evidence that M. tuberculosis forms biofilms in vitro, which harbour drug-tolerant populations of cells that persist despite exposure to high levels of antibiotics. Their in vitro formation is common amongst isolates throughout the world suggesting that their formation is relevant to TB propagation or persistence (Pang et al. 2012).
Whilst the long-term persistence of M. tuberculosis in the presence of the host immune system and chemotherapy bears a clear similarity to the chronic infections of biofilm-forming pathogens, it remains unclear whether this pathogen forms biofilms in the host (Ojha and Hatfull 2012). The non-tuberculous mycobacterial species, Mycobacterium ulcerans and Mycobacterium avium, do, however, colonize the host as biofilms (Carter et al. 2003; Marsollier et al. 2005). Even though M. tuberculosis may be restricted to the phagosome during early stages of infection, many of the bacteria likely experience an extracellular environment later on, when lesions contain liquefied caseum and when patients are highly infective (Ojha and Hatfull 2012). This is further exemplified by the observations that TB lesions, including open cavities, contain numerous extracellular bacilli growing in multicellular structures (Canetti 1955) and by the presence of surviving micro-colonies in infected guinea pigs after drug treatment, located around the acellular rim in the granulomas (Lenaerts et al. 2007).
Microbial community behaviour is complex and can involve many genetic loci, particularly those that encode extracellular factors that promote surface colonization or cell-to-cell contact (Barnhart and Chapman 2006). Several bacteria, including Escherichia coli and Salmonella spp., are known to produce extracellular fibres called curli pili that are important mediators in biofilm formation (Barnhart and Chapman 2006). Although it was previously believed that mycobacteria did not produce pili, Alteri et al. (2007) provided compelling evidence for the existence of M. tuberculosis curli-like pili (MTP). Fibre analysis identified the Rv3312A (mtp) gene product as a component of MTP. These researchers also provided initial genetic evidence for the role of mtp in piliation by generating Δmtp mutants of M. tuberculosis H37Rv and CDC1551 that lacked detectable MTP (Alteri et al. 2007).
In the present study, we sought to provide further genetic evidence for the requirement of mtp in pili production by generating Δmtp mutant and mtp-complemented strains of a M. tuberculosis clinical isolate; evaluating mtp gene expression in these strains by quantitative PCR; and assessing their ability to form pili by transmission electron microscopy. Furthermore, we aimed to determine whether these surface appendages contribute to M. tuberculosis biofilm formation in vitro and evaluate mtp gene expression of wild-type cells growing in biofilms versus planktonic conditions.
Materials and methods
Bacterial strains and growth conditions
Mycobacterium tuberculosis V9124, a drug-susceptible clinical isolate of the F15/LAM4/KZN (KZN) family, isolated from the Tugela Ferry (KwaZulu-Natal, South Africa), together with its mutant and complemented strains, were propagated on solid Middlebrook 7H10 or 7H11 medium (Difco), containing 10 % (v/v) oleic acid-albumin-dextrose-catalase (OADC) supplement (Becton–Dickinson) and 0.5 % (v/v) glycerol (Sigma) or for planktonic growth in liquid Middlebrook 7H9 medium (Difco), supplemented with 10 % (v/v) OADC, 0.5 % (v/v) glycerol, and 0.05 % (v/v) Tween 80 (Sigma). E. coli strains were cultured in Luria–Bertani (LB) broth or agar (Difco). The concentrations of antibiotics used were 150 μg ml−1 hygromycin (Roche Applied Sciences) and 30 μg ml−1 kanamycin (Sigma) for mycobacterial strains, and 150 μg ml−1 hygromycin and 40 μg ml−1 kanamycin for E. coli.
Bioinformatics of the mtp gene and its translation product
The mtp gene sequence and locus was obtained from the TubercuList website (Lew et al. 2011). The mtp gene sequence of the clinical isolate used in this study was confirmed by PCR amplification and sequencing (N. Naidoo, S. Ramsugit, and M. Pillay; submitted). Secondary structure and topology of the corresponding amino acid sequence were determined using the PredictProtein software (Rost et al. 2004). Signal peptide was predicted using SignalP 4.0 server (Petersen et al. 2011). Protein hydrophobicity was predicted using a Kyte-Doolittle Hydropathy Plot (Kyte and Doolittle 1982).
Construction of mtp deletion mutant (Δmtp::γδsacBhygγδ)
An allelic exchange substrate (AES) designed to replace the mtp gene with a hygromycin-resistance (hyg R)-sacB cassette was kindly donated by W. R. Jacobs, Jr., AECOM. This had been generated by PCR amplification of the sequences flanking the left and right regions of the M. tuberculosis mtp gene using the primer pairs Mtp-LL and Mtp-LR, and Mtp-RL and Mtp-RR, respectively (Table 1). Subsequently, upstream and downstream arms were digested with BstAPI and Van91I, respectively, and cloned into Van91I-digested pYUB1471 vector arms. The plasmid pYUB1471 allows one step generation of AES and contains a hyg R-sacB cassette, lambda phage cos sites, and a unique PacI site to clone into temperature-sensitive mycobacteriophage phAE159 shuttle phasmid. The resulting AES was digested with PacI and ligated with PacI-digested phAE159, in vitro packaged using a MaxPlax packaging extract (Epicentre Biotechnologies), and transduced in the E. coli HB101 strain (Invitrogen Life Technologies) for construction of shuttle phasmids. The donated phasmids were electroporated into Mycobacterium smegmatis mc2155 (kind gift from W. R. Jacobs, Jr., AECOM) and incubated at 30 °C for phage propagation (Bardarov et al. 2002). Specialized transduction of the M. tuberculosis V9124 wild-type strain was performed as described by Bardarov et al. (2002). A 188-bp region of the mtp gene was subsequently deleted and replaced with a γδres-sacB-hyg-γδres cassette, and hygromycin was used for the selection of mutants.
PCR screening of deletion mutants
Genomic DNA of the putative deletion mutants was isolated using the NaCl-cetyl trimethylammonium bromide method (Ausubel et al. 1989). The 312-bp mtp gene was amplified using primer pairs Mtp-1F and Mtp-1R, to generate a 370-bp product, as well as Mtp-2F and Mtp-2R, to generate a 583-bp product (Table 1), to confirm the loss of the mtp gene in the mutant compared to the wild-type strain. The PCR mix contained: 1X buffer with 1.5 mM MgCl2, 250 μM dNTPs, 0.2 μM of each primer, 2 U Taq polymerase (Roche Applied Sciences), and 100 ng DNA per 25 μl of reaction mixture. PCR amplification was performed under the following conditions: 95 °C for 1 min; 40 cycles of 95 °C for 30 s, 58 °C for 1 min, and 72 °C for 30 s; and a final cycle with an extension time of 5 min at 72 °C. All standard PCR amplifications were carried out on the GeneAmp PCR System 9700 (Applied Biosystems) and amplicons were resolved by electrophoresis in a 1.5 % (w/v) agarose (Seakem) gel, containing ethidium bromide, and visualized in a UV trans-illuminator (Syngene G-Box, Vacutec).
Replacement of the mtp gene by the hyg R-sacB cassette was further confirmed by amplification of a 300-bp portion of the hyg R cassette using 2 μM of each primer Hyg-F and Hyg-R (Table 1), 1X buffer with 500 μM MgCl2, 200 μM dNTPs, 2.5 U Taq polymerase, and 100 ng DNA per 50 μl of reaction mixture. The thermal cycling conditions included: 94 °C for 5 min; 25 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s; and a final extension of 7 min at 72 °C.
Southern hybridization analysis of the deletion mutant
A 783-bp fragment downstream of the mtp gene was PCR-amplified using primers SH-F and SH-R (Table 1). The amplification reaction mixture (25 μl) contained: 1X buffer with 1.5 mM MgCl2, 250 μM dNTPs, 0.2 μM of each primer, 1 U Taq polymerase, and 100 ng wild-type DNA. The cycling parameters were 95 °C for 10 min; followed by 40 cycles of 95 °C for 20 s, 50 °C for 1 min, and 72 °C for 1 min; and finally 5 min at 72 °C. The PCR product was labeled with Horse Radish Peroxidase (Amersham) and hybridized to NheI-digested wild-type and mutant genomic DNA and detected using enhanced chemiluminescence (Amersham; Millipore). The designed probe was expected to detect a 2,744-bp fragment for the wild-type and a 6,357-bp fragment for the deletion mutant.
Complementation of the Δmtp strain
The mtp gene was cloned into the pMV261 vector (Stover et al. 1991) for complementation into the Δmtp mutant strain as follows. A 436-bp fragment containing the mtp gene was amplified using PCR primers Mtp-261F and Mtp-261R (Table 1) that have BglII and HindIII restriction sites incorporated, respectively. The PCR mix contained: 1X buffer with 1.5 mM MgCl2, 250 μM dNTPs, 0.2 μM of each primer, 1 U Taq polymerase, and 100 ng wild-type DNA per 25 μl of total reaction volume. PCR amplification was performed under the following conditions: 95 °C for 10 min; 40 cycles of 95 °C for 20 s, 59 °C for 1 min, and 72 °C for 30 s; and a final cycle with an extension time of 5 min at 72 °C. The gel-purified (QIAquick Gel Extraction Kit, Qiagen) PCR product and vector were double-digested with restriction enzymes BglII and HindIII, and BamHI and HindIII (Thermo Scientific), respectively. The digested PCR product was cloned downstream of the mycobacterial hsp60 promoter of the digested vector to generate pMV261::mtp. This ligation mixture was transformed into chemically-competent E. coli JM109 cells (kind gift from D. T. Claiborne, Emory University) and transformed cells selected by plating on LB agar plates containing kanamycin. Plasmid DNA was isolated from transformed colonies with the PureLink HQ Mini Plasmid Purification Kit (Invitrogen Life Technologies) and screened for the 436-bp mtp insert by PCR. Electrocompetent cells of the Δmtp mutant strain were prepared and electrotransformed with the identified pMV261::mtp construct (Larsen et al. 2007). Transformants were selected on kanamycin-containing agar plates.
PCR screening and sequencing of mtp-complemented strain
Plasmid DNA was isolated from transformed M. tuberculosis colonies (Larsen et al. 2007) and the 436-bp sequence was once again amplified by PCR for confirmatory purposes. To ensure that no mutations were produced during amplification, and thereby confirming the integrity of the construct, PCR products were sequenced (Inqaba Biotec, Pretoria, South Africa) using the same primer set, and Chromas Pro and Bioedit software were used to analyze the chromatograms and perform multiple sequence alignment.
IS6110, a M. tuberculosis complex-specific insertion sequence element, was amplified using primers INS-1 and INS-2 (Table 1) (van Soolingen et al. 1991) to confirm that the derived Δmtp mutant and mtp-complemented strains were M. tuberculosis, which was also confirmed by Ziehl-Neelsen staining.
RNA extraction and quantitative real-time PCR (qPCR)
In order to provide further evidence for the generation of the Δmtp mutant and mtp-complemented strains, mtp gene expression in the wild-type, mutant, and complemented strains were analysed by qPCR. After the cultures were grown aerobically on 7H11 agar plates for 7 weeks at 37 °C, total RNA was extracted with TRIzol reagent (Invitrogen Life Technologies), precipitated with isopropanol overnight, resuspended in Ultra-pure DNase/RNase-free water (Bioline), and the concentration and purity were determined using a NanoDrop (Thermo Scientific). Total RNA (200 ng) was reverse-transcribed using the High-Capacity cDNA Reverse Transcription kit (Roche Applied Sciences) according to the manufacturer’s protocol. Real-time PCR analysis was carried out on the CFX96 Real-Time PCR Detection System (Bio-Rad). Five microlitres of the cDNA reaction was used in a 25 μl reaction, containing 1X TaqMan Universal PCR Master Mix (Applied Biosystems), 4 mM primer Mtp-AnyF, 4 mM primer Mtp-AnyR, and 2.5 U μl−1 mtp probe (Table 1) with a 5’-FAM and 3′-minor groove binder (MGB) non-fluorescent quencher (NFQ). The amplification protocol involved 95 °C for 10 min and 50 cycles of: 95 °C for 15 s and 60 °C for 1 min. Expression was normalized against the expression of 16S rRNA housekeeping gene, using primers 16S-AnyF and 16S-AnyR and the 16S probe (Table 1). Each reaction was repeated with three independent RNA samples. Negative/no template controls were included in each experiment. Normalized expression (using the ΔΔCq method) was determined using the CFX Manager Software and results were expressed as fold change in expression of the target gene in the mutant and complemented strains relative to the wild-type expression.
Transmission electron microscopy (TEM)
The wild-type, mutant, and complemented strains that were grown on 7H11 agar for 9 weeks were fixed in 4 % paraformaldehyde overnight at room temperature. After heat-killing at 80 °C for 1 h, a drop of bacterial suspension was placed onto 200 square mesh copper grids (Agar Scientific) and removed after 5 min. The samples were negatively-stained with 2 % aqueous uranyl acetate for 5 min and rinsed with a drop of sterile water. The dried samples were viewed using a JEOL 1010 transmission electron microscope at an accelerating voltage of 100 kV.
Biofilm growth conditions
Using a modification of published protocols (Ojha et al. 2008); the wild-type, mutant, and complemented strains were cultured for biofilm growth in 6-well polystyrene plates (Porvair). Cells from 7H9 medium-aerated cultures (absorbance at 600 nm [A600] = 1) were diluted 1:100 (v/v) in Sauton’s medium without detergent and 2 ml added to each well in triplicate. The plates were wrapped twice in Parafilm and incubated at 37 °C in 5 % CO2 for 5 weeks without shaking. Serial dilutions of the bacterial inocula were plated in triplicate onto 7H11 plates to confirm that similar numbers of viable cells were included for all three strains in the assay. Plates were assessed visually after 5 weeks for biofilm formation.
Quantification of biofilm mass
Crystal violet staining for the quantification of biofilm mass was conducted by modifying previously described methods (Carter et al. 2003; Pang et al. 2012). Medium was aspirated by pipetting. The remaining biofilm mass was dried in a biosafety cabinet and incubated with 0.5 ml of 1 % crystal violet solution for 10 min. The wells were washed thrice with water and dried again. Two and a half millilitres of 95 % ethanol was added to each well for 10 min and the absorbance of the resulting biofilm cell-associated dye was measured at A600 on a spectrophotometer (Biochrom WPA Lightwave II, Labotec), using identically-treated planktonically-grown cultures to blank each sample.
qPCR analysis
Total RNA from 5 week old planktonic and biofilm wild-type cultures was extracted and qPCR used to evaluate mtp expression during biofilm growth relative to that during planktonic growth, as described above.
Results and discussion
Organization of the mtp gene locus
A schematic illustrating the chromosomal organization of the mtp (Rv3312A) gene in the wild-type and in a mtp deletion mutant strain is shown in Fig. 1a. The mtp gene is located between genes involved in intermediary metabolism (add, deoA, and cdd) and a conserved mycobacterial gene of unknown function (Rv3312c), an organization notably different to the operons of pili biogenesis genes observed in other curli piliated bacteria. In E. coli and Salmonella spp., curli pili operons include the csgBA and csgDEFG (Hammar et al. 1995) and the agfBA and agfDEFG operons (Collinson et al. 1996), respectively, with both species requiring at least six proteins for the production of curli. However, pili genes are not always arranged in clusters and there is a possibility that the MTP biogenesis genes are located distantly on the chromosome (Alteri 2005).

Construction of the mtp deletion mutant by allelic replacement and its confirmation. a A schematic illustrating the chromosomal organization of the wild-type (upper) and the Δmtp mutant (lower) strains, in which the mtp gene has been disrupted by a hyg R-sacB cassette, and expected sizes following restriction with NheI and Southern hybridization. (b, c) Confirmation of the mutant by PCR. In b the mtp gene was amplified using primers Mtp-1F and Mtp-1R, as well as Mtp-2F and Mtp-2R, yielding products of 370-bp and 583-bp in the wild-type (Lanes 2 and 3), but absent in the mutant (Lanes 4 and 5). In c the hygromycin-resistance cassette was detected using primers Hyg-F and Hyg-R, yielding a 300-bp product in the mutant (Lane 3), that is absent in the wild-type (Lane 2). d Southern blot showing digested genomic DNA of the wild-type (Lane 1) and mutant (Lane 2), probed downstream of the mtp gene. The location of the probe is also illustrated in the schematic in (a)
The absence of any sortase-encoding genes in M. tuberculosis, that are normally required for pili assembly in Gram-positive species (Paterson and Mitchell 2004); the lack of a β-sheet structure typically seen in curlins and amyloid fibres (Table 2); a general limited knowledge on curli pili in Gram-positive organisms; coupled with the complexity of the cell wall architecture of M. tuberculosis, point to a unique system of pili biogenesis in this organism (Alteri et al. 2007).
Generation of the M. tuberculosis Δmtp::γδsacBhygγδ (Δmtp) mutant and complemented strain
In order to confirm the role of the mtp gene in pili production, a precise deletion mutant of mtp was generated by deleting 188-bp of the 312-bp mtp sequence using specialized transduction (Bardarov et al. 2002) (Fig. 1a). This deletion of about 60 % of the sequence of the mtp gene is expected to suffice in hindering functioning of the translation product, as protein sequence analysis using a combination of bioinformatics tools showed that the deleted region includes important functional domains (Table 2). This includes the transmembrane region that is necessary for spanning the protein across a biological membrane; the signal peptide region necessary for secretion of the protein; 51.85 % of the amino acids contributing to the highly folded/looped nature of curlins; 67.86 % of the amino acids associated with protein binding; a major portion of the identified hydrophilic region of the protein which would be exposed on the surface of the protein; and the 2 cysteine residues at the C-terminus important for disulphide formation and protein stability (Alteri et al. 2007). The genotype of the Δmtp mutant strain was confirmed by PCR (Fig. 1b and 1c) and Southern hybridization (Fig. 1d).
To ensure that the mutant phenotype resulted from the loss of mtp and not secondary mutations that may have arisen during its construction, complementation of the mutant strain was performed. An extrachromosomal plasmid with wild-type mtp was used to complement the mutant. The genotype of the complemented strain was confirmed by PCR and sequencing (data not shown).
qPCR analysis showed no amplification of mtp in the mutant samples, thereby confirming production of a Δmtp mutant strain (Table 3). Expression of the reference gene indicates that the lack of mtp expression was not due to degradation of the isolated RNA samples. In contrast, there was a 51-fold increase in mtp expression in the complemented strain, relative to the wild-type (Table 3). This over-expression of the mtp gene was expected since the mtp gene expression in the complemented strain was driven by the constitutively-expressed hsp60 promoter, and to a lesser extent the 3–5 copy number of the plasmid.
Evidence that the mtp gene is involved in pili production
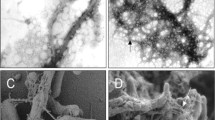
To demonstrate the phenotypic consequence of the mtp gene knockout and the subsequently-derived complemented strain, transmission electron microscopy was employed to directly view negatively-stained pili. The wild-type strain was shown to be piliated (Fig. 2a). Only 16 of the 20 individual wild-type cells produced pili-like structures, possibly due to the absence of signals and environmental cues required for pili expression during in vitro growth (Alteri 2005).

The requirement of mtp in piliation. Transmission electron micrographs of negatively-stained M. tuberculosis V9124 wild-type (a), Δmtp mutant (b), and mtp-complemented (c) strains. Black arrows point to pili fibres. Scale bars 0.2 μm
Expression was, however, much higher than the 10–20 and 10–15 % piliated bacteria reported by Alteri et al. (2007) and Velayati et al. (2012), respectively. The higher percentage piliation seen in this study highlights the strain- and condition-specific expression of curli, and is most likely largely due to the older cultures used here (9 weeks, as opposed to 2–3 weeks), as nutrient limitation has been shown to stimulate curli gene expression in organisms such as Salmonella typhimurium (Gerstel and Romling 2001). This occurrence was confirmed by wild-type mtp gene expression analysis by qPCR of a 5 week versus 15 week culture in which mtp expression was 6.35-fold higher in the older culture. This observation indicates that mtp are possible genetic determinants involved in the long-term dormancy and persistence of M. tuberculosis.
The pili structures identified by electron microscopy were hair-like and aggregated to form a network/mesh of fibres that extended out of the cell surface to varying degrees. They also seemed to facilitate inter-bacterial adherence between these organisms, suggesting that mtp are possibly genetic loci involved in cell aggregation and biofilm formation.
Analysis of the Δmtp mutant strain demonstrated an absence of surface appendages. Every cell examined (n = 30) lacked pili (Fig. 2b). TEM of the complemented strains demonstrated that piliation was clearly restored (Fig. 2c). Copious amounts of pili fibres were expressed by every cell examined (n = 20). These results support those of the transcript analysis and, taken together, confirm that the mtp gene is required for pili formation in M. tuberculosis.
Pili contribute to M. tuberculosis biofilm formation
To determine whether MTP contribute to biofilm formation, we assessed the ability of the wild-type, Δmtp mutant, and mtp-complemented strains to form biofilms in vitro. The wild-type strain produced a biofilm phenotype (Fig. 3a). Cells were aggregated to each other forming several sediments and a pellicle at the liquid-air interface. In the absence of MTP, in the pili-deficient mutant strain, biofilm formation was clearly defective. There was no liquid surface growth and minimal attachment to the container surface in comparison to the wild-type (Fig. 3b). Colony forming unit (CFU) determinations confirmed that a similar number of bacterial cells was included for the wild-type and mutant biofilm assays (8.3 × 107 and 7.8 × 107 CFU’s per ml, respectively). This indicated that the defect in biofilm formation in the mutant wells was not due to inoculation of low viability mutant inoculums. Furthermore, subsequent growth of the biofilm-grown mutant cells under planktonic conditions resulted in restored growth, similar to the wild-type cells in a planktonic state, indicating that the lack of biofilm formation in the mutant was not due to growth defects or a loss of viability.

The requirement of M. tuberculosis pili (MTP) in biofilm formation. M. tuberculosis V9124 wild-type (a), Δmtp mutant (b), and mtp-complemented (c) strains were inoculated into Sauton’s media in 6-well plates and incubated for 5 weeks at 37 °C and 5 % CO2 without shaking
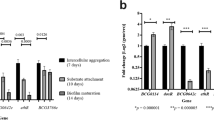
Crystal violet staining for biofilm mass determination showed a 68.43 % reduction in biofilm mass in the mutant compared to the parental strain (P = 0.002) (Fig. 4). Complementation resulted in a restoration of the wild-type biofilm phenotype (P = 0.022) (Figs. 3c, 4). The sediments seen in the pili over-expressing mtp-complemented strain were much larger than in the wild-type, suggesting the role of pili in facilitating aggregation and biofilm maturation.

Crystal violet quantification of biofilm mass of the wild-type, mutant, and complemented strains. Results are expressed as mean absorbance values at 600 nm, derived from triplicate samples. Standard deviations are also shown. The experimental data were analyzed by one-way analysis of variance (ANOVA) using SPSS 21.0 statistical software; F(2,6) = 21.969, P < 0.05
To determine whether pili expression is enhanced during biofilm growth, we assessed mtp gene expression in the V9124 wild-type strain cultured under planktonic and biofilm growth conditions by qPCR. It was, however, found that mtp expression in cells of the biofilm were similar to those growing planktonically.
Whilst mtp expression is not up-regulated or specific to the biofilm lifestyle, the results of this study, nevertheless, indicate that pili are important contributors to the community lifestyle of M. tuberculosis. Their role most likely would be to facilitate surface attachment and cell-to-cell contact, thus contributing to the structural integrity of the biofilm. Alteri (2005) showed that statically grown M. tuberculosis H37Ra adhered to glass coverslips and each other using pilus-like structures. Their possible role in aggregation to other bacterial cells was previously noted by Velayati et al. (2012) using atomic force microscopy of clinical isolates of M. tuberculosis. This cellular aggregation may be attributed to the highly hydrophobic nature of the MTP protein (Alteri 2005).
Gao et al. (2004) suggested the role of mtp in another complex M. tuberculosis aggregative phenotype known as cording, a phenomenon associated with virulence in a variety of models (Middlebrook et al. 1947). Using microarray technology, they showed that the mtp gene was consistently expressed at higher levels in H37Rv, a virulent, cording strain of M. tuberculosis, compared with H37Ra, an avirulent, non-cording strain derived from the same original patient isolate.
The finding by Alteri et al. (2007) that MTP are expressed in vivo and bind strongly to the eukaryotic extracellular matrix protein laminin supports the notion that pili could also be actively-engaged in surface attachment and biofilm formation in vivo (Islam et al. 2012).
Concluding remarks
Since biofilms are important reservoirs for the development of drug-tolerant persistent organisms, the identification of the role of MTP in biofilm formation in this study indicates that they may represent an important target for the development of new, more effective drugs against TB. These drugs, termed curlicides (Cegelski et al. 2009), that block the formation of curli, would disrupt biofilm formation, and when used in combination with current anti-TB drugs may enable a much shorter and more effective treatment regimen. Further studies are underway to identify the role of MTP in other aspects of TB pathogenesis.
References
Alteri CJ (2005) Novel pili of Mycobacterium tuberculosis. Ph.D. Thesis, The University of Arizona
Alteri CJ, Xicohténcatl-Cortes J, Hess S, Caballero-Olín G, Girón JA, Friedman RL (2007) Mycobacterium tuberculosis produces pili during human infection. Proc Natl Acad Sci USA 104:5145–5150
Ausubel FM, Brent R, Kingston RE, Moore DD, Smith JA, Seidman JG, Struhl K (1989) Current protocols in molecular biology. Greene Publishing and Wiley Interscience, New York
Bardarov S, Bardarov S Jr, Pavelka MS Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR Jr (2002) Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017
Barnhart MM, Chapman MR (2006) Curli biogenesis and function. Annu Rev Microbiol 60:131–147
Canetti G (1955) Tubercle Bacillus in the pulmonary lesion of man: histobacteriology and its bearing on the therapy of pulmonary tuberculosis. Springer, New York
Carter G, Wu M, Drummond DC, Bermudez LE (2003) Characterization of biofilm formation by clinical isolates of Mycobacterium avium. J Med Microbiol 52:747–752
Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Åberg V, Walker JN, Seed PC, Almqvist F, Chapman MR, Hultgren SJ (2009) Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat Chem Biol 5:913–919
Collinson SK, Clouthier SC, Doran JL, Banser PA, Kay WW (1996) Salmonella enteritidis agfBAC operon encoding thin, aggregative fimbriae. J Bacteriol 178:662–667
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322
Gao Q, Kripke K, Arinc Z, Voskuil M, Small P (2004) Comparative expression studies of a complex phenotype: cord formation in Mycobacterium tuberculosis. Tuberculosis 84:188–196
Gerstel U, Rőmling U (2001) Oxygen tension and nutrient starvation are major signals that regulate agfD promoter activity and expression of the multicellular morphotype in Salmonella typhimurium. Environ Microbiol 3:638–648
Hammar M, Arnqvist A, Bian Z, Olsén A, Normark S (1995) Expression of two csg operons is required for production of fibronectin- and congo red-binding curli polymers in Escherichia coli K-12. Mol Microbiol 18:661–670
Islam MS, Richards JP, Ojha AK (2012) Targeting drug tolerance in mycobacteria: a perspective from mycobacterial biofilms. Expert Rev Anti Infect Ther 10:1055–1066
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132
Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR Jr (2007) Genetic manipulation of Mycobacterium tuberculosis. Curr Protoc Microbiol 6:10A.2
Lenaerts AJ, Hoff D, Aly S, Ehlers S, Andries K, Cantarero L, Orme IM, Basaraba RJ (2007) Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother 51:3338–3345
Lew JM, Kapopoulou A, Jones LM, Cole ST (2011) TubercuList- 10 years after. Tuberculosis (Edinb) 91:1–7
Marsollier L, Aubry J, Coutanceau E, André JP, Small PL, Milon G, Legras P, Guadagnini S, Carbonnelle B, Cole ST (2005) Colonization of the salivary glands of Naucoris cimicoides by Mycobacterium ulcerans requires host plasmatocytes and a macrolide toxin, mycolactone. Cell Microbiol 7:935–943
Middlebrook G, Dubos RJ, Pierce C (1947) Virulence and morphological characteristics of mammalian tubercle bacilli. J Exp Med 86:175–184
O’Toole G, Kaplan HB, Kolter R (2000) Biofilm formation as microbial development. Annu Rev Microbiol 54:49–79
Ojha AK, Hatfull GF (2012) Biofilms of Mycobacterium tuberculosis: new perspectives of an old pathogen. In: Cardona P (ed) Understanding tuberculosis—deciphering the secret life of the bacilli. Intech Open Access Publisher, Reijek, pp 181–192
Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, Guerardel Y, Alahari A, Kremer L, Jacobs WR Jr, Hatfull GF (2008) Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol Microbiol 69:164–174
Pang JM, Layre E, Sweet L, Sherrid A, Moody DB, Ojha A, Sherman DR (2012) The polyketide Pks1 contributes to biofilm formation in Mycobacterium tuberculosis. J Bacteriol 194:715–721
Paterson GK, Mitchell TJ (2004) The biology of Gram-positive sortase enzymes. Trends Microbiol 12:89–95
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Rost B, Yachdav G, Liu J (2004) The PredictProtein Server. Nucleic Acids Res 32(Web Server issue):W321–W326
Sasindran SJ, Torrelles JB (2011) Mycobacterium tuberculosis infection and inflammation: what is beneficial for the host and for the bacterium? Front Microbiol 2:2
Spigelman M, Ma Z (2004) Mycobacterium tuberculosis: new tricks for an old bug. Expert Rev Anti Infect Ther 2:467–469
Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR Jr, Bloom BR (1991) New use of BCG for recombinant vaccines. Nature 351:456–460
van Soolingen D, Hermans PW, de Haas PE, Soll DR, van Embden JD (1991) Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J Clin Microbiol 29:2578–2586
Velayati AA, Farnia P, Masjedi MR (2012) Pili in totally drug resistant Mycobacterium tuberculosis (TDR-TB). Int J Myco 1:57–58
World Health Organization (2012) Global tuberculosis report 2012. http://apps.who.int/iris/bitstream/10665/75938/1/9789241564502_eng.pdf. Accessed 6 March 2013
Acknowledgments
We thank Mr Mhlengi Vella Ncube (UKZN) for his contribution to the generation of the Δmtp mutant; Ms Charissa Naidoo (UKZN) for help with the statistical analysis; and the National Research Foundation (NRF), SA, Medical Research Council (MRC), SA, and College of Health Sciences (CHS), UKZN, for financial support. Mr S. Ramsugit gratefully acknowledges scholarship from the NRF and Canon Collins Trust.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramsugit, S., Guma, S., Pillay, B. et al. Pili contribute to biofilm formation in vitro in Mycobacterium tuberculosis . Antonie van Leeuwenhoek 104, 725–735 (2013). https://doi.org/10.1007/s10482-013-9981-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-013-9981-6