
Abstract
Arid zones cover over 30 % of the Earth’s continental surface. In order to better understand the role of microbes in this type of harsh environment, we isolated and characterized the bacteriophages from samples of the surface sand of the Mesquite Flats region via electron microscopy and DNA sequencing of a select number of cloned phage DNAs. An electron microscopic analysis of the recovered virus-like particles revealed at least 11 apparently different morphotypes sharing structural characteristics of the Caudoviridae family of tailed phages. We found that 36 % of the sequences contained no significant identity (e-value >10−3) with sequences in the databases. Pilot sequencing of cloned 16S rRNA genes identified Bacteroidetes and Proteobacteria as the major bacterial groups present in this severe environment. The majority of the 16S rDNA sequences from the total (uncultured) bacterial population displayed ≤96 % identity to 16S rRNA genes in the database, suggesting an unexplored bacterial population likely adapted to a desert environment. In addition, we also isolated and identified 38 cultivable bacterial strains, the majority of which belonged to the genus Bacillus. Mitomycin-C treatment of the cultivable bacteria demonstrated that the vast majority (84 %) contained at least one SOS-inducible prophage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the recognition of increasing global climate changes, desertification is becoming a problem that has attracted attention from a number of sources. The consequences of desertification can be dramatic in terms of the loss of cultivatable land, threatening the economy and health of neighbouring populations, in addition to airborne transport of desert sand to distant locations (Kellogg and Griffin 2006). Arid zones cover over 30 % of the Earth’s continental surface, and are defined as regions that receive less than 150 mm of precipitation per year. As water is essential for life on Earth, deserts are considered as an extreme environment, with concomitant reduced biodiversity.
However, arid zones have been found to contain a large diversity of animals, plants and microbial eukaryotes and prokaryotes that have evolved the capacity to live in these harsh environments (Chesson et al. 2004; Lewis and Lewis 2005). In deserts, as in other environments, microorganisms are key components in the proper functioning of the ecosystem, such as the maintenance of soil physical structure (West 1990).
In order to better understand the role of microbes in this type of environment, it is important to increase our knowledge of arid region microbial diversity. The bacterial diversity of different Antarctic cold desert mineral soils, the Tataouine region of the Sahara desert, soil crusts in the Sonoran desert, and the Atacama desert have been examined (Smith et al. 2006; Chanal et al. 2006; Nagy et al. 2005; Drees et al. 2006). However, only recently have the viruses of bacteria (called bacteriophages) been identified in hot desert sand samples (Prigent et al. 2005; Fierer et al. 2007; Prestel et al. 2008), as well as in cold desert soils (Williamson et al. 2007). Bacteriophages are the most abundant living entities on Earth (Hendrix et al. 2003) and have been discovered in a variety of diverse extreme environments (Le Romancer et al. 2007). The majority of available data concerning bacteriophage diversity in environmental samples comes from aquatic, or relatively humid, ecosystems. In marine and fresh-water ecosystems, bacteriophages can be found at densities of up to 108 virus-like particles (VLPs) per millilitre, and are, on average, tenfold more abundant than their bacterial hosts (Bergh et al. 1989). In soils, fewer studies concerning viruses have been published. In a rhizosphere in Scotland, numerous viral particles have been observed (Swanson et al. 2009) and direct counts have estimated the density of bacteriophages as 1.5 × 108/g (Ashelford et al. 2003). A crucial rapid bacteria-phage coevolution has been suggested to explain the structure, population dynamics, and the function of soil microbial communities (Gomez and Buckling 2011). The effects of bacteriophages on bacterial community function in soils is also reinforced by the prevalence of lysogeny, and thus including prophage gene expression plus superinfection immunity, as a phage lifestyle (Ghosh et al. 2008; Williamson et al. 2007).
Because of their abundance and capacity to affect the growth and diversity of bacteria, phages play a key role in the control of bacterial populations and thus in ecosystem function (Weinbauer 2004). Moreover, the relationships between bacteriophages and their hosts can often be more complex than simple cell killing. Examples include selective advantages conferred by temperate bacteriophages to their hosts, such as the bor function of λ that provides protection to Escherichia coli against the human innate immune system (Barondess and Beckwith 1995). Another striking example is the mutualist symbiosis between pea aphids and the bacterium Hamiltonella defensa that itself contains the temperate Podiviridae phage APSE-2, whose genome encodes a toxin that allows the pea aphid to resist the attack of the wasp Aphidius ervi (Moran et al. 2005). The role of phage-mediated horizontal gene transfer (transduction) on bacterial evolution has also been well documented (Canchaya et al. 2003). Comparisons of the bacteriophages in hot, versus cold and/or temperate soils, are much less numerous (Williamson et al. 2007; Williamson 2011).
In order to obtain more information on phages and their hosts in hot desert soils, we report here an exploration of the bacterial and bacteriophage diversity of a hot surface desert sand sample taken from the Mesquite Flats region of the Death Valley desert in California (USA). The oldest rocks found in this desert have been dated to the Precambrian at approximately 1.8 billion years old. Temperatures in Death Valley can range from up to 54 °C in the day in summer, to below freezing at night in winter. Death Valley attains temperatures that are among the hottest on Earth. The hottest temperature recorded in the USA was 56.7 °C at Furnace Creek (Death Valley) on July 10, 1913. Death Valley receives less than 50 mm of rain annually, and years without any rain have been recorded (data from the Death Valley National Park, http://www.nps.gov/deva).
We have previously characterized the morphology of bacteriophages from samples of the surface sand of the Sahara and Namib deserts (Prigent et al. 2005; Prestel et al. 2008) via electron microscopy, and potential genome diversity using pulsed-field gel electrophoresis. Here, in addition to the above criteria, phage diversity was also examined by random amplification and cloning of phage DNA, followed by sequencing of a select number of clones. Sequencing of random cloned 16S rRNA genes identified Bacteroidetes and Proteobacteria as the major bacterial groups present in this sand sample. In addition, we identified 38 cultivable bacterial strains, the majority of which belonged to the genus Bacillus. Mitomycin-C treatment of the cultivable bacteria demonstrated that the vast majority (84 %) contained at least one SOS-inducible prophage.
Materials and methods
Surface (0–3 cm depth) sand samples were aseptically collected (by direct scooping) from the surface of a dune of the Death Valley desert in the Mesquite Flats region (GPS coordinates: 36.45 N; 117.15 W) into three sterile 50 ml polypropylene conical centrifuge tubes (a kind gift of E. Marguet and P. Forterre, Université Paris-Sud) in December 2005. Samples were returned to the laboratory within 1 week, and then processed.
Procedure for the isolation of bacteriophages
The procedure for the isolation of bacteriophages from the desert sand was adapted from that of Prigent et al. (2005). Five grams of sand were resuspended in two different bacteriological culture media: 10 ml LB medium (Lennox L: 1 % Bacto-Tryptone, 0.5 % Bacto-Yeast Extract, 1 % NaCl, NaOH [3 × 10−3 M]) or 10 ml ¼ TS medium (0.25× Tryptic Soy broth, Difco). Samples were sonicated for 15 s at 33 % duty cycle at a maximum output of 4 using a microprobe on a Branson model 450 Sonifier, followed by two different incubation times (1 or 3 h) at 30 °C with 150 rpm shaking. The samples were then subjected to centrifugation at 6,000×g 10 min, 4 °C and the supernatant fluids were collected. Each pellet (bacteria and sand particles) was resuspended in 10 ml of the same medium containing freshly-prepared 1 μg/ml mitomycin-C (Sigma-Aldrich), and incubated for 20 min at 30 °C with 150 rpm shaking. After two washing steps to remove the mitomycin-C, the samples were resuspended and incubated for 2 h at 30 °C in 10 ml of fresh medium. In order to extract the bacteriophages, the cells, debris and sand were centrifuged for 10 min at 6,000 g and 4 °C. The supernatant fluids were removed and filtered through a 0.22 μm pore-size filter (Millipore). The bacteriophages were concentrated by ultracentrifugation at 100,000 g for 3 h at 4 °C (Beckman TL-100 ultracentrifuge, TLS 100.4 rotor), and after removal of the supernatant fluid, the phage pellets were overlayed with 50 μl of Mu buffer (20 mM Tris–HCl pH 7.5, 200 mM NaCl, 20 mM MgSO4, 1 mM CaCl2, 0.1 % (w/v) gelatin) at 4 °C overnight. The pellet was then gently resuspended and stored at 4 °C.
To determine the presence of temperate bacteriophages in cultivable bacteria, mid-log growth phase cultures of each bacterial strain were subjected to mitomycin-C treatment, and phages partially purified, as described above.
Electron microscopy
Formvar, carbon-coated copper electron microscopy grids (400 mesh) were overlaid with 10 μl of concentrated bacteriophage suspensions for 5 min, negatively stained with 1 % (w/v) phospho-tungstic acid for 1 min and then allowed to air dry. The bacteriophages were observed using an EM 205 Philips electron microscope (80 kV microscope accelerating voltage). For each sample, phages from two grids were examined to distinguish specific morphotype groups of the major observable Caudoviridae VLPs. Lower and higher numbers are presented that represent stringent versus relaxed criteria for each VLP to be assigned to a specific morphotype.
Total DNA extraction from sand
The DNA extraction protocol was adapted from that of Zhou et al. (1996). Five grams of sand were added to 13.5 ml extraction buffer (100 mM Tris–HCl pH 8, 100 mM NaEDTA, 100 mM Na2HPO4, 1.5 M NaCl, 1 % (w/v) CTAB) containing 1 μg/ml self-digested pronase (final concentration) and incubated for 2 h at 37 °C with 200 rpm horizontal shaking. Then, 1.5 ml 20 % (w/v) SDS was added and a supplementary 2 h incubation at 65 °C was performed with the samples mixed by inversion every 20 min during this step. After centrifugation at 6,000×g for 10 min at room temperature, the supernatant fluid was collected. The pellet was extracted two more times with 4.5 ml extraction buffer plus 0.5 ml 20 % (w/v) SDS, mixed by vortex for 10 s, and incubated at 65 °C for 10 min. The nucleic acids were extracted through the addition of an equal volume of chloroform/isoamyl alcohol (24:1), and precipitated by the addition of 0.6 volume of isopropanol for 1 h at room temperature. After a 20 min centrifugation at 16,000×g, the nucleic acid pellet was washed with 70 % ethanol. The pellet was allowed to solubilize in 1/10 TE (1 mM Tris–HCl, pH 8; 0.1 mM NaEDTA) at 4 °C for 24 h and stored at −20 °C until use (Tolias and DuBow 1985).
Isolation of cultivable bacteria
After suspension and sonication of the sand samples in culture media, 1 ml of the suspension was spread on petri dishes containing LB-agar (for the sand dispersed in LB medium), or ¼ TS-agar (for the sand dispersed in ¼ TS medium). Plates were then incubated at 30 °C for up to 96 h. Based on colony morphology, 38 different strains were isolated through two serial streak plates. The bacterial clones were grown in liquid media and stored at –80 °C in the presence of 15 % (v/v) glycerol.
Cultivable bacterial genomic DNA extraction
Isolated strains were cultivated overnight in LB medium. The cells in 3 ml of the culture were harvested by centrifugation and the cell pellet was resuspended in 250 μl P1 buffer, and then 250 μl buffer P2 was added (Qiaprep Spin kit, Qiagen). The nucleic acids were extracted twice by the addition of an equal volume of phenol/chloroform/isoamyl alcohol (24/24/1) and the DNA was precipitated by the addition of 2.5 volumes of 100 % ethanol, followed by a 1 h incubation at −80 °C. The DNA precipitate was collected by centrifugation at 20,000×g for 20 min at 4 °C, washed in 70 % ethanol and resuspended in 50 μl sterile 1/10 TE.
Phage DNA extraction
Prior to phage DNA extraction, free RNA and DNA present in the phage preparations were removed by a 1 h incubation at 37 °C with 1 μg/ml DNAseI and 1 μg/ml RNAse A. Bacteriophage genomic DNA was then extracted from the virions by the addition of 1 volume of lysing buffer (50 mM Tris–HCl pH 7,5, 2 mM NaEDTA, 1 % (w/v) SDS, 13 % (w/v) sucrose) and incubation for 10 min at 65 °C. The DNA was purified by phenol/chloroform/isoamyl alcohol (25/24/1) extraction, followed by ether extraction and ethanol precipitation as described above.
Bacteriophage DNA random amplification and cloning
Bacteriophage DNA was amplified using the protocol described by Bohlander et al. (1992) and Wang et al. (2002). A first round of random amplification was carried out using primer A (5′-GTTTCCCAGTCACGATCNNNNNNNNN-3′) (Operon), 8 units of Sequenase enzyme, 1× sequenase buffer (USB Europe, Staufen, Germany) and 3 mM dNTPs (Fermentas). Subsequently, the result of this first amplification was used as the template for 30 cycles of PCR amplification with primer E (5′-GTTTCCCAGTAGGTCTC-3′) using the following program: 30 s at 94 °C, 30 s at 40 °C, 30 s at 50 °C, 2 min at 72 °C and 2.5 units of High Fidelity PCR enzyme Mix (Fermentas). When necessary, the amplified fragments were purified using the MinElute PCR purification Kit (Qiagen), cloned into the pSmart LCKan vector as recommended by the manufacturer (Lucigen), and transformed into electrocompetent E. coli DH10B cells.
16S rDNA amplification and cloning
16S rDNA was amplified from the sand-extracted DNA using universal primers 8F and 1492R (Crump et al. 1999), and the following protocol: 2 μM primer, 0,4 mM dNTPs (Fermentas), 1× Thermopol buffer (Fermentas), and 2 U High Fidelity PCR enzyme mix (Fermentas). PCR condition were as follows: 5 min at 95 °C, followed by 30 cycles of 1 min at 95 °C, 30 s at 51 °C, 2 min at 72 °C, and a 7 min final elongation at 72 °C. The MinElute PCR Purification Kit (Qiagen) was used to purify the amplified fragments. The PSmart LCKan (Lucigen) kit, with E. coli DH10B competent cells, were used as cloning vector and host.
For the isolated cultivable bacteria, the 16S rRNA genes were amplified using the same primers and the same PCR conditions, except that GoTaq polymerase (Promega) was used in order to perform TA cloning of the amplified fragments in the PDrive vector (Qiagen) and transformed into electro-competent E. coli DH10B cells.
Sequencing and DNA sequence analyses
Clones with plasmids containing 16S rRNA genes, or bacteriophage DNA fragments, were sequenced on both strands by GENOME EXPRESS (Cogenics, Meylan, France) using the pSMART primers SL1, SR2 (Lucigen) or the T7/Sp6 primers for the PDrive vector. Sequences of plasmid origin were removed by hand. The remaining sequences were checked for the presence of chimeras using the program Bellerophon (Huber et al. 2004) available online at the Greengenes (http://greengenes.lbl.gov/) web site. Sequences were compared against the GenBank, RDP 2, Greengenes and ssurRNA databases using BLASTn for 16S rRNA gene analyses. For phage DNA analyses, BLASTX searches were performed using virus protein and nr protein and the comparison was considered significant if a similar sequence had an e-value ≤0.001.
The 16S rDNA sequences identified here are available in the NCBI database under accession numbers EU362130 to EU362184, while the phage DNA sequences are available in the NCBI database under accession numbers ET190212 to ET190260.
Phylogenetic analysis
The 16S rRNA gene sequences obtained were aligned with sequence representatives of each close relative by BLAST analysis (for which the accession number is indicated in brackets), using the CLUSTALW algorithm (Chenna et al. 2003). Alignments were manually verified by comparing the homologous positions of related sequences. The phylogenetic trees were calculated with the software package MEGA version 3 (Kumar et al. 2004), using the neighbour-joining (NJ) method (Saitou and Nei 1987) and the Kimura-2 model (Kimura 1980). Bootstrap analyses were based on 1,000 replicates, and only values >50 % are indicated.
Results
Bacteriophage electronmicroscopic analysis
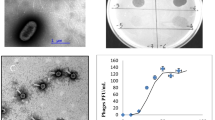
The sand samples from the Mesquite Flats site of Death Valley were examined in order to search for the presence of bacteriophages in desert environments. Incubation of the sand in two different types of bacterial culture media, in the absence and presence of mitomycin-C, was performed in order to recover intracellular phages (SOS-inducible prophages and phages within “pseudolysogens”) (Armon 2011; Leroy et al. 2008; Prestel et al. 2008; Prigent et al. 2005). Phages were recovered and then collected by ultracentrifugation. An electron microscopic analysis of the recovered VLPs revealed at least 11 apparently different morphotypes (Fig. 1). In addition, all of these VLPs shared structural characteristics of the Caudovirales order. Only two short tailed VLPs, with icosahedral capsids and apparently belonging to the Podoviridae, were observed (Fig. 1b, d). The remaining VLPs appeared to be equally distributed between the Siphoviridae (non contractile flexible tails) and Myoviridae (contractile tails) phage morphotypes. The capsid size of the Myoviridae and Siphoviridae VLPs ranged from 50 to 100 nm, with tail lengths ranging from 100 to 700 nm for the Siphoviridae, and from 100 to 500 nm for the Myoviridae type bacteriophages.

Examples of phage particle isolates from the Death Valley desert sample. Phage particles were isolated as described in the Materials and methods section and examined with a Philips 205 electron microscope on Formvar carbon-coated copper grids after negative staining with phosphotungstate. Arrows indicate the structures of VLPs
The relative abundance and diversity of the VLPs from the different incubation conditions are shown in Table 1. The VLPs were only observed in significant numbers after mitomycin-C treatment, suggesting that the active phage population is mostly present as SOS-inducible prophages (Williamson et al. 2007; Ghosh et al. 2008). After a 1 h incubation of the sand in each medium, three different VLP morphotypes were detected, two of which were isolated from the sand sample incubated in ¼ TS medium (Fig. 1a, b; Table 1), while the other VLP morphotype was found to be present after incubation of the sand sample in LB medium. Interestingly, this bacteriophage is characterized by the presence of an apparent ribbon at the extremity of the tail, rather than individual fibers (Fig. 1c). This peculiar morphology has been previously observed in other desert sand samples (Prestel et al. 2012). After 3 h of incubation, the number of different VLP morphotypes observed was increased from 3 to 11 VLP morphotypes (Table 1).
VLP DNA cloning and sequence
In order to extend the data concerning the Death Valley phage community, a molecular approach, using random PCR DNA amplification, cloning, and sample sequencing, was undertaken. The VLPs from the phage preparations after 1 and 3 h incubations in the four media used (LB or ¼ TS ± mitomycin-C addition) were pooled and their nucleic acids extracted, amplified and the resultant fragments subsequently cloned. Selected clones, containing inserts of >400 bp, were chosen and sequenced. We found that 36 % of the sequences contained no significant identity (e-value >10−3) with sequences in the databases, 28 % possessed homologies with sequences of bacterial origin, and 36 % yielded significant similarities with known bacteriophage sequences (Fig. 2). Additionally, 82 % of the sequences were found to possess a GC content of <50 %, with 27 % having a GC content of <40 %. In the “unknown” category, a large proportion were found to contain sequences with very low GC content. Except for two sequences that displayed homologies with sequences of bacteriophages able to infect Proteobacteria, the analysis suggests the presence of many sequences belonging to phages infecting low GC gram positive bacteria. The rest of the sequences have significant homologies with phages infecting proteobacteria often present in soils (Table 2).

Distribution of the phage sequences as a function of their best blastX hit. Black areas represent the proportion of sequences with no significant homology to sequences in the databases. Dark grey areas represent the proportion of sequences with significant homology to known bacterial sequences and the light grey area represents the proportion of sequences with significant homology to phage sequences in the databases
Bacterial 16S rDNA analysis
In order to identify the abundant bacterial community members present in the sand of the Mesquite Flats samples, total DNA was extracted from the sand and 16S rRNA genes were PCR amplified using universal bacterial primers. After purification, cloning, and DNA sequencing of selected random full length 16S rRNA genes, the sequences were analysed against different 16S rDNA databases. The major detected bacterial group was found to belong to the Bacteroidetes, followed by members of the Proteobacteria. The remaining bacterial 16S rDNA sequences were dispersed among several other bacterial groups. Strikingly, the majority of 16S genes displayed less than 97 % identity (for example: 91 % similar in the case of DV203 and a sequenced, yet uncultured, Gemmatimonadetes) with the 16S rDNA sequences available in the databases, suggesting an unexplored bacterial diversity adapted to this environment (Fig. 3a).

Phylogenetic trees of the 16S rDNA sequences identified from the uncultivable and cultivable bacteria of the Death Valley sand samples. a Tree constructed with all the obtained sequences except those belonging to the genus Bacillus. b Tree drawn with the related Bacillus species sequences. Trees were calculated as described in Materials and methods. Bacterial names in bold correspond to 16S rDNA sequences from cultivable bacterial strains, while bacterial strains identified from total sand-extracted DNA are also underlined. The letter “ϕ” indicates the detection of a mitomycin-C-inducible prophage from a cultivable isolated strain
Isolation and identification of cultivable bacteria
Cultivable bacteria that could grow under laboratory conditions in the two media used for incubation of the sand samples were isolated and characterized. Approximately equal numbers of CFUs were found between the two different media used (LB or ¼ TS), suggesting the presence of approximately 5 × 103 cfu/g of sand. After re-streaking of the morphologically different colonies obtained, all strains isolated on LB were found to be able to grow on ¼ TS and vice versa. On the basis of criteria such as colony color, morphology (edge shape, mucoid or not), and size of the colonies, 38 of the most likely different colonies were chosen for subsequent characterisation.
In order to identify each isolated strain, genomic DNA was extracted, the 16S rRNA genes amplified by PCR and cloned into the pDrive vector. The results of the sequencing of the different rDNA clones are presented in Fig. 3a and b. The vast majority (95 %) of the strains were found to be Gram positive and most belong to the phylum Firmicutes, with one Planomicrobium and one strain whose 16S rRNA gene displayed 99 % sequence identity with that of Saccharibacillus kuerlensis, a recently isolated bacterium from a desert soil in China (Yang et al. 2009). The majority of the Bacillus species detected were similar to B. mojavensis, B. vallismortis, B. sonorensis and B. niabiensis that have been isolated and identified from other desert samples.
The remaining clones belong to the Actinobacteria and are distributed between the Arthrobacter and Microbacterium genera. Only two Gram negative cultivable bacterial species were isolated from the Mesquite Flats sand samples used in this study. Both are members of the Proteobacteria, and are representatives of the Pseudomonas (γ-Proteobacteria) and Erwinia (β-Proteobacteria) genera.
Induction and observation of cultivable bacterial prophages
The cultivable bacteria from the Death Valley Mesquite Flats sand samples were examined for the presence of temperate bacteriophages after mitomycin-C treatment of an LB culture of each bacterial isolate, grown to mid-log phase. Bacteria were removed by filtration and the filtrates were subjected to ultracentrifugation in order to collect any phages that may have been produced. The concentrated phage preparations were then examined for the presence of DNA on agarose gels. No phages were observable by electron microscopy in the preparations in which no discernable DNA was detected (data not shown). However, in all preparations containing discernable DNA, after examination by electron microscopy, VLPs were found in significant quantities. The vast majority of the bacterial isolates (32/38) were found to contain at least one temperate bacteriophage inducible by mitomycin-C. Except for three isolates, all of the remaining Bacillus clones were found to produce mitomycin-C inducible bacteriophages. The most frequently encountered morphotype was a Myoviridae type bacteriophage with a capsid diameter of approximately 50 nm and a contractile tail approximately 330 nm in length (Fig. 4; ϕDV4, ϕDV44, ϕDV48, ϕDV16). A Siphoviridae morphotype phage was produced from bacterial clones DV49 and DV19, with a long thin tail of 330 nm, along with a Myoviridae type phage with a small head (45 nm) and a tail of 220 nm in length (ϕDV5). Strain DV1 produced a Siphoviridae phage morphotype (Fig. 4; ϕDV1) with a large head (100 nm) and a very long tail of 450 nm. In the group of cultivable Actinobacteria, only one type of phage morphotype was observed to be inducible from the different Microbacterium strains, a Siphoviridae type phage with a 70 nm capsid and a short tail of 120 nm in length. The phages designated ϕDV40, ϕDV42 and ϕDV46, induced from the Arthrobacter species, were composed of two different morphotypes, a Siphoviridae with a long tail (350 nm) and a Myoviridae phage (Fig. 4).

Examples of phage particles isolated from each phylum of cultivable bacteria after induction with mitomycin-C. Same legend as in Fig. 1
Discussion
The microbial biodiversity of hot desert ecosystems has attracted increasing interest in recent years (Drees et al. 2006; Chanal et al. 2006; Nagy et al. 2005; Dunbar et al. 2002; Fancello et al. 2013; Neilson et al. 2012) including analyses of the bacteriophages from this type of environment (Fierer et al. 2007; Prigent et al. 2005; Prestel et al. 2008; Prestel et al. 2012). A metagenomic analysis of phages extracted from three different soil environments (prairie, desert and rain forest) (Fierer et al. 2007) suggests that similar phage types can be found among all three soil ecosystems. However, the phage DNA sequences from each soil were different and significantly dissimilar from phages present in aquatic environments, suggesting an enormous genetic diversity among the 3 soils examined. However, no electron microscopic observations of the phage morphotypes were reported.
Although we did not observe VLPs or phage DNAs in the absence of an incubation step, we were able to visualize bacteriophages after 1 or 3 h incubations of the sand in culture medium, as previously reported (Prigent et al. 2005; Prestel et al. 2008). An examination of phage diversity in a cold desert soil ecosystem revealed that the extracellular phages were abundant and likely preserved due to the cold temperatures (Williamson 2011). While free viruses are likely present in low abundance in the hot Death Valley sand, it is known that dry, mineral sand particles can irreversibly adsorb and inactivate extracellular VLPs and render their extraction too harsh to maintain their morphological integrity (Rossi 1994; Williamson et al. 2003). The sonication and incubation steps were included to allow phage-infected bacteria, pseudolysogens, (Elasri et al. 2000) and SOS-inducible phages to complete a lytic cycle, with incubations limited to 1 or 3 h to minimize skewing of the population and thus more accurately reflect the diversity of the indigenous sand microbial community (Prigent et al. 2005; Prestel et al. 2008). In addition to LB broth, diluted TS broth was used to augment phage recovery by allowing bacterial metabolism of a plant-based medium and to minimize nutrient shock to the cells (Reasoner and Geldreich 1985). The addition of mitomycin-C is frequently employed to induce the lytic cycle of SOS-inducible prophages (Williamson et al. 2007). However, this antibacterial compound is unstable at temperatures above 4 °C, while still retaining its cytotoxic effect (Velpandian et al. 2005). Thus, we limited the exposure time to 30 min using freshly-prepared mitomycin-C, followed by a wash step to remove the mitomycin-C plus subsequent incubation, to allow SOS-inducible temperate phages to complete a lytic cycle (Prigent et al. 2005; Prestel et al. 2008) to reduce skewing the production of phages to those from resistant bacteria. Lysogeny is thought to be a viral survival strategy at low host densities and may also confer advantages to the host, including immunity to superinfection by homoimmune phages (Ackermann and DuBow 1987). More studies will be required to determine the proportion of lysogeny in soil bacteria (Stopar et al. 2004; Williamson et al. 2007). The greater phage diversity observed when incubating sediment in different culture media and in the absence and presence of mitomycin-C suggests that intracellular phages may be a significant fraction of the viral community (Leroy et al. 2008), particularly in soil and sediment environments (Ghosh et al. 2008).
The bacteriophages observed here share the structural characteristics of the Caudovirales (Ackermann and DuBow 2000). It is difficult to assess how many different bacteriophages are present using electron microscopy, as sample preparation can cause alterations in phage morphology (Williamson et al. 2012). However, phage diversity at the morphological level appears to be quite large for this type of harsh, low biomass environment.
To gain insights into the genetic diversity of the phages recovered from the sand, we amplified fragments of phage DNA and sequenced a number of randomly selected clones. A significant portion (36 %) of the sequences bore no significant identity to sequences in the databases, a finding in line with that reported for phages isolated from soil (Fierer et al. 2007) or aquatic (Breitbart et al. 2004) environments. Based on an analysis of marine phages, from 65 to 73 % of the phage DNA sequences had no significant identity (cutoff value e <0.001) with sequences in the databases, a situation also observed with viral sequences from human feces (59 % of the sequences) (Breitbart et al. 2003). Approximately 90 % of sequences obtained from marine phages (Angly et al. 2006) displayed no significant homologies with sequences in the databases when a cut off value of e ≤10−5 was used.
Several facts can explain these results. First, there are relatively few annotated bacteriophage sequences in the databases compared with their estimated total biodiversity. Moreover, bacteriophage sequences can be incorrectly annotated as bacterial chromosomal sequences when, in reality, they represent unrecognized prophages or cryptic phage fragment regions (Fouts 2006). The sequences we found that possessed homologies with bacteriophage sequences included phage-related proteins such as virion structural proteins and enzymes linked to the replication of phage DNA. The largest number of sequence identities observed in our sample were with those from phages isolated from Firmicutes. However, as phage genomes are often constructed with modules, these homologies may be of limited use in precise phage identification (Romero et al. 2004).
Cloning and sequencing of selected 16S rDNA genes from the total DNA extracted from the sand revealed that the majority represented members of the phylum Bacteroidetes, not including the cultivable bacteria. We also found members of the Proteobacteria, which are common environmental bacteria, though more frequently observed in temperate and aqueous environments. Members of the Proteobacteria have been found in surveys of bacterial biodiversity in desert sand samples (Nagy et al. 2005; Chanal et al. 2006). It is worth noting that a majority of the 16S rDNA sequences from the total (uncultured) bacterial population displayed ≤96 % identity with 16S rDNA genes in the database. Thus, the rDNA genes we characterised suggest an unexplored bacterial population likely adapted to a desert environment.
When cultivable bacteria were examined, we found that a majority belonged to the Firmicutes (76 %), and in particular to members of the Bacilli, and to the Actinobacteria, and specifically to members of the Arthrobacter/Microbacteria genera. In order to document the presence of SOS-inducible prophages in the desert sand, bacterial prophage induction by mitomycin-C of the cultivable bacteria was explored. The vast majority (84 %) of the bacterial isolates were found to contain at least one mitomycin-C inducible prophage. It is essential to remember that surface desert sand is a very mobile soil, frequently on the move via wind (aeolian) action. Thus, a small (1–3 cm) layer of sand, such as sampled here, will not long attenuate UV irradiation due to particle movement and rotation leading to eventual direct bacterial and/or phage exposure to sunlight. It is interesting to speculate that bacteria, in addition to resistant spores, may exist within biofilms on the surface of the sand particles. Biolfilms are known to provide protection against UV radiation and desiccation (Donlan 2002; Elasri and Miller 1999; Elasri et al. 2000). Gommeaux et al. (2010) microscopically examined sand grain particle surfaces from the Sahara desert, and found evidence of bacteria (via Syto 9 staining) on the surface of individual sand grains. It is unclear if the individual bacteria on the sand grains were encased in extracellular biopolymers (i.e. a unicellular biofilm). It is important to note here that the silts and clays (i.e. the smaller sand mineral particles) are perhaps where minute biofilms may be located as particle-associated dried organic matter (such as from desert plant rhizospheres or decaying dead animals, for example).
This work suggests that both bacteria and their phages can survive the harsh conditions of surface sand found in the Mesquite Flats region of the Death Valley desert. Many of the phage DNA sequences yielded no significant homology with sequences in the databases, while many of the bacterial 16S rDNA sequences were sufficiently divergent from known bacterial rRNA sequences as to likely represent novel bacterial species. Their mechanisms of survival are not, as yet, clearly understood. These results also suggest that temperate phages may play an important role in desert sand viral processes, perhaps during favorable growth conditions (Armon 2011; Williamson 2011). The increasing awareness of the microbial biodiversity observed in deserts suggests that further study of the bacterial and phage ecology of this extensive terrestrial ecosystem will be vital.
References
Ackermann HW, DuBow MS (1987) Viruses of procaryotes. CRC Press, Cleveland
Ackermann HW, DuBow MS (2000) Myorividae, siphoviridae, podoviridae. In: Van Regenmotel MHV, Bishop DHL, Carstens EB, Estes MK, Lemon SM, Maniloff J, Mayo MA, Mcgeoch DJ, Pringle CR, Wickner RB (eds) Virus taxonomy classification of viruses seventh report of the international committee on taxonomy of viruses. Academic Press, San Diego, pp 69–109
Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, Chan AM, Haynes M, Kelley S, Liu H, Mahaffy JM, Mueller JE, Nulton J, Olson R, Parsons R, Rayhawk S, Suttle CA, Rohwer F (2006) The marine viriomes of four oceanic regions. PLoS Biol 4:2121–2131
Armon R (2011) Soil bacteria and bacteriophages. In: Witzany G (ed) Biocommunication in soil microorganisms, soil biology 23. Springer-Verlag, Berlin, pp 67–112
Ashelford KE, Day MJ, Fry JC (2003) Elevated abundance of bacteriophage infesting bacteria in soil. Appl Environ Microbiol 69:285–289
Barondess JJ, Beckwith J (1995) Bor gene of phage lambda, involved in serum resistance, encodes a widely conserved outer membrane lipoprotein. J Bacteriol 177:1247–1253
Bergh O, Borsheim KY, Bratbak G, Heldal M (1989) High abundance of viruses found in aquatic environments. Nature 340:467–468
Bohlander SK, Espinosa R 3rd, Le Beau MM, Rowley JD, Diaz MO (1992) A method for the rapid sequence-independent amplification of microdissected chromosomal material. Genomics 13:1322–1324
Breitbart M, Hewson I, Felts B, Mahaffy JM, Nulton J, Salamon P, Rohwer F (2003) Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol 185:6220–6223
Breitbart M, Felts B, Kelley S, Mahaffy JM, Nulton J, Salamon P, Rohwer F (2004) Diversity and population structure of a near-shore marine-sediment viral community. Proc Biol Sci 271:565–574
Canchaya C, Fournous G, Chibani-chennoufi S, Dillmann ML, Brussow H (2003) Phage as agents of lateral gene transfer. Curr Opin Microbiol 6:417–424
Chanal A, Chapon V, Benzerara K, Barakat M, Christen R, Achouak W, Barras F, Heulin T (2006) The desert of Tataouine: an extreme environment that hosts a wide diversity of microorganisms and radiotolerant bacteria. Environ Microbiol 8:514–525
Chenna R, Sugawara H, Koite T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003) Multiple sequence alignment with the clustal series of programs. Nucleic Acids Res 31:3497–3500
Chesson P, Gebauer RL, Schwinning S, Huntly N, Wiegand K, Ernest MS, Sher A, Novoplansky A, Weltzin JF (2004) Resource pulses, species interactions, and diversity maintenance in arid and semi-arid environments. Oecologia 141:236–253
Crump BC, Armbrust VE, Baross JA (1999) Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia River, its estuary, and the adjacent coastal ocean. Appl Environ Microbiol 65:3192–3204
Donlan R (2002) Biofilms: microbial life on surfaces. Emerg Infect Dis 8:881–890
Drees KP, Neilson JW, Betancourt JL, Quade J, Henderson DA, Pryor BM, Maier RM (2006) Bacterial community structure in the hyperarid core of the Atacama desert, Chile. Appl Environ Microbiol 72:7902–7908
Dunbar J, Barns SM, Ticknor LO, Kuske CR (2002) Empirical and theoretical bacterial diversity in four Arizona soils. Appl Environ Microbiol 68:3035–3045
Elasri MO, Miller RV (1999) Study of the response of a biofilm bacterial community to UV radiation. Appl Environ Microbiol 65:2025–2031
Elasri MO, Reid T, Hutchens S, Miller ME (2000) Response of a Pseudomonas aeruginosa biofilm community to DNA-damaging chemical agents. FEMS Microbiol Ecol 33:21–25
Fancello L, Trape S, Robert C, Boyer M, Popgeorgiev N, Raoult D, Desnues C (2013) Viruses in the desert: a metagenomic survey of viral communities in four perennial ponds of the Mauritanian Sahara. ISME J 7:359–369
Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R, Robeson M, Edwards RA, Felts B, Rayhawk S, Knight R, Rohwer F, Jackson RB (2007) Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl Environ Microbiol 73:7059–7066
Fouts DE (2006) Phage finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res 34:5839–5851
Ghosh D, Roy K, Williamson KE, White DC, Wommack KE, Sublette KL, Radosevich M (2008) Prevalence of lysogeny among soil bacteria and presence of 16S rRNA and trzN genes in viral-community DNA. Appl Environ Microbiol 74:495–502
Gomez P, Buckling A (2011) Bacteria-phage antagonistic coevolution in soil. Science 332:106–109
Gommeaux M, Barakat M, Montagnac G, Christen R, Guyot F, Heulin T (2010) Mineral and bacterial diversities of desert sand grains from south to east Morocco. Geomicrobiol J 27:76–92
Hendrix RW, Hatfull GF, Smith MC (2003) Bacteriophages with tails: chasing their origins and evolution. Res Microbiol 154:253–257
Huber T, Faulkner G, Hugenholtz P (2004) Bellerophon; a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317–2319
Kellogg CA, Griffin DW (2006) Aerobiology and the global transport of desert dust. Trends Ecol Evol 21:638–644
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Le Romancer M, Gaillard M, Geslin C, Prieur D (2007) Viruses in extreme environments. Rev Environ Sci Biotechnol 6:17–31
Leroy M, Prigent M, Dutertre M, Confalonieri F, DuBow MS (2008) Bacteriophage morphotype and genome diversity in Seine River sediment. Freshw Biol 53:1176–1185
Lewis LA, Lewis PO (2005) Unearthing the molecular phylodiversity of desert soil green algae (Chlorophyta). Syst Biol 54:936–947
Moran NA, Degnan PH, Santos SR, Dunbar HE, Ochman H (2005) The players in a mutualistic symbiosis: insects, bacteria, viruses and virulence genes. Proc Natl Acad Sci USA 102:16919–16926
Nagy ML, Perez A, Garcia-Pichel F (2005) The prokaryotic diversity of biological soil crusts in the Sonoran desert (Organ Pipe Cactus National Monument, AZ). FEMS Microbiol Ecol 54:233–245
Neilson JW, Quade J, Ortiz M, Nelson WM, Legatzki A, Tian F, LaComb M, Betancourt JL, Wing RA, Soderlund CA, Maier RM (2012) Life at the hyperarid margin: novel bacterial diversity in arid soils of the Atacama desert, Chile. Extremophiles 16:553–566
Prestel E, Salamitou S, DuBow MS (2008) An examination of the bacteriophages and bacteria of the Namib desert. J Microbiol 46:364–372
Prestel E, Regeard C, Andrews J, Oger P, DuBow MS (2012) A novel bacteriophage morphotype with a ribbon-like structure at the tail extremity. Res J Microbiol 7:75–81
Prigent M, Leroy M, Confalonieri F, Dutertre M, DuBow MS (2005) A diversity of bacteriophage forms and genomes can be isolated from the surface sands of the Sahara desert. Extremophiles 9:289–296
Reasoner DJ, Geldreich EE (1985) A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol 49:1–7
Romero P, Lopez R, Garcia E (2004) Genomic organization and molecular analysis of the inducible prophage EJ-1, a mosaic myovirus from an atypical pneumococcus. Virology 322:239–252
Rossi P (1994) Advances in biological tracer techniques for hydrology and hydrogeology using bacteriophages. Ph.D. thesis, University of Neuchâtel, Neuchâtel, Switzerland
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Smith JJ, Tow LA, Stafford W, Cary C, Cowan DA (2006) Bacterial diversity in three different Antarctic cold desert mineral soils. Microb Ecol 51:413–421
Stopar D, Cerne A, Zigman M, Poljak-Prijatelj M, Turk V (2004) Viral abundance and a high proportion of lysogens suggest that viruses are important members of the microbial community in the Gulf of Trieste. Microb Ecol 47:1–8
Swanson MM, Fraser G, Daniell TJ, Torrance L, Gregory PJ, Taliansky M (2009) Viruses in soils: morphological diversity and abundance in the rhizosphere. Ann Appl Biol 155:51–60
Tolias PP, DuBow MS (1985) The cloning and characterization of the bacteriophage D108 regulatory DNA-binding protein Ner. EMBO J 4:3031–3037
Velpandian T, Saluja V, Ravi AK, Kumari SS, Mathur R, Ranjan N, Ghose S (2005) Evaluation of the stability of extemporaneously prepared ophthalmic formulation of mitomycin C. J Occul Pharmacol Ther 21:217–222
Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, DeRisi JL (2002) Microarray-based detection and genotyping of viral pathogens. Proc Natl Acad Sci USA 99:15687–15692
Weinbauer MG (2004) Ecology of prokaryotic viruses. FEMS Microbio Rev 28:127–181
West NE (1990) Structure and function of microphytic soils crust in wildland ecosystems of arid to semi-arid regions. Adv Ecol Res 20:179–223
Williamson KE (2011) Soil phage ecology: abundance, distribution, and interaction with bacterial hosts. In: Witzany G (ed) Biocommunication in soil microorganisms, soil biology 23. Springer-Verlag, Berlin, pp 113–136
Williamson KE, Wommack KE, Radosevich M (2003) Sampling natural viral communities from soil for culture-independent analyses. Appl Environ Microbiol 69:6628–6633
Williamson KE, Radosevich M, Smith DW, Wommack KE (2007) Incidence of lysogeny within temperate and extreme soil environments. Environ Microbiol 9:2563–2574
Williamson KE, Helton RR, Wommack KE (2012) Bias in bacteriophage morphological classification by transmission electron microscopy due to breakage or loss of tail structures. Microsc Res Tech 75:452–457
Yang SY, Liu H, Liu R, Zhang KY, Lai R (2009) Saccharibacillus kuerlensis sp. nov., isolated from a desert soil. Int J Sys Evol Microbiol 59:953–957
Zhou J, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Envrion Microbiol 62:316–322
Acknowledgments
The authors would like to thank Jeril Degrouard and Danielle Jaillard for their help with the electron microscopy, Evelyne Marguet and Patrick Forterre (Université Paris-Sud, Orsay, France) for their generosity in providing the sand samples from the Mesquite Flats site of Death Valley, and the Editor and Reviewers for their terrific comments, criticisms and suggestions. This work was supported by the AQUAPHAGE program of the Agence Nationale de la Recherche (ANR), France, and the Centre National de la Recherche Scientifique (CNRS, France).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Prestel, E., Regeard, C., Salamitou, S. et al. The bacteria and bacteriophages from a Mesquite Flats site of the Death Valley desert. Antonie van Leeuwenhoek 103, 1329–1341 (2013). https://doi.org/10.1007/s10482-013-9914-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-013-9914-4