
Abstract
The use of genetically modified (GM) plants still raises concerns about their environmental impact. The present study aimed to evaluate the possible effects of GM maize, in comparison to the parental line, on the structure and abundance of microbial communities in the rhizosphere. Moreover, the effect of soil type was addressed. For this purpose, the bacterial and fungal communities associated with the rhizosphere of GM plants were compared by culture-independent methodologies to the near-isogenic parental line. Two different soils and three stages of plant development in two different periods of the year were included. As evidenced by principal components analysis (PCA) of the PCR-DGGE profiles of evaluated community, clear differences occurred in these rhizosphere communities between soils and the periods of the year that maize was cultivated. However, there were no discernible effects of the GM lines as compared to the parental line. For all microbial communities evaluated, soil type and the period of the year that the maize was cultivated were the main factors that influenced their structures. No differences were observed in the abundances of total bacteria between the rhizospheres of GM and parental plant lines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Globally, the area cultivated with genetically modified (GM) crops is growing fast. It now represents 148 million hectares, corresponding to 10 % of the total cultivated area around the world. The number of countries that plant GM crops is increasing, and in particular in developing countries GM plant cultivation is growing impressively (James 2010). There are clear advantages in the utilization of GM plants in a world with limited resources and a rapidly increasing population, one of them being the reduced need for application of broad-spectrum chemical pesticides such as in the case of maize carrying Bacillus thuringiensis (Bt) insect toxin genes (McLaren and Copping 2011; Gruber et al. 2012).
Bt-maize has become a favorite anti-insect crop worldwide. In particular the gene for the insecticidal crystal protein that is toxic to Lepidoptera such as the European corn borer has been widely used (Feng et al. 2011). This has led to concern that, besides its intended effect on the target insect, there might be unintended effects. However, in spite of previous studies with respect to possible effects on the soil (micro) biota, there is as yet no consensus about such effects (Gruber et al. 2012). A likely route via which the soil microbiota can be affected might be secretion of the Bt toxin through root exudation. Thus, specific communities might be selected that are able to use this protein as a carbon source, leading to a shift in the structure of the microbial communities associated with the rhizosphere (Dunfield and Germida 2004). Indeed, some groups have reported no differences between the microbial communities in soils planted with GM versus non-GM maize crops (Tan et al. 2010, 2011). In contrast, other studies found some effects related to use of GM crops, such as a modification of mycorrhizal fungal colonization patterns, alteration of culturable bacteria and fungi and a change in the active fungal communities (Motavalli et al. 2004; Castaldini et al. 2005; Hanulla et al. 2012). Yu and co-workers (Yu et al. 2011) commented that the potential effects of Bt crops on soil microorganisms are difficult to track in soil. Unfortunately, these studies were developed in temperate climate regions, in spite of the fact that the major GM-producing countries nowadays are located in tropical regions (James 2010). The few studies found in literature that have evaluated the possible effects of transgenic plants on microbial communities in tropical soils did not use Bt-plants (Andreote et al. 2008, 2009).
Thus, one may conclude that the impact of the introduction of Bt GM crops on the soil microbiota, including bacteria and fungi, is still unclear in particular in tropical climate zones (Carpenter 2011; Yu et al. 2011). Bacteria and fungi are the dominant organisms in soil and represent critical components of biogeochemical cycles. They are responsible for the breakdown of organic matter, mineralization and fixation of nutrients, control of plant disease and amelioration of soil structure. They can provide indicators of soil health (Bruinsma et al. 2003; Raybould et al. 2011). In addition to the potential effects of GM plants on the soil microbiota, their ecological success may also depend on dynamic relationships with microbial communities in the soil (Raybould et al. 2011).
Taking into consideration the importance of the bacterial and fungal communities in the rhizosphere, the present work aimed to evaluate, by culture-independent methodologies, the impact of Bt maize, as compared to that of a non-transgenic line, in a tropical environment. Furthermore, the actinobacterial community was evaluated in the light of the prevalence of this bacterial group on maize roots in a tropical soil (Chauhan et al. 2011). For this, two GM maize lines were cultivated in two different soil types. The putative impacts of these plants were monitored during three plant developmental stages. The plants were cultivated twice, i.e. during two periods of the year: the first one (known as “Safra”) which happens in the rainy period (on average around 300 mm of precipitation/second semester/harvest in August) and the second one (known as “Safrinha”) in a drier period of the year (on average around 60 mm of precipitation/first semester/harvest in February).
Materials and methods
Maize cultivars and experimental conditions
The field experiment was carried out at “EMBRAPA Milho e Sorgo”, Sete Lagoas, Minas Gerais, Brazil, located at latitude 19°28′S and longitude 44°15′W, at a height of 732 m. The local climate is of the savannah type, with a mean temperature in the coldest month above 18 °C, according to the Köppen classification. Two different types of soil found in “EMBRAPA Milho e Sorgo” were used, denoted Cerrado (dark-red distrophic latosol with a clayey texture) and Várzea (low humus eutrophic gley soil with a clayey texture). The characteristics of both soils are presented in Table S1. They are representative of soils commonly cropped with maize in Brazil. Two hybrid Bt-maize genotypes were used: (i) the Guardian hybrid (Monsanto), a GM maize cultivar which represents transformation event MON810 genotype 30F35Y (expressing the insecticidal Bt-toxin Cry1Ab from B. thuringiensis), and (ii) the Herculex hybrid (Pioneer), which was recently approved for commercialization in Brazil. This cultivar represents the transformation event TC1507 genotype 30F35H (expressing the insecticidal Bt-toxin Cry1F from B. thuringiensis). The near-isogenic parental line of both GM maize cultivars was used as the control. The plants were cultivated during the traditional period for maize cultivation in Brazil, known as “Safra” (second semester/harvest in August—rainy period of the year) and also during a non-traditional period for maize cultivation, known as “Safrinha” (first semester/harvest in February—dry period of the year). The experimental set-up consisted of completely randomized fourfold replicated 5 × 5 m plots containing four rows of 5 m length with spaces of 0.8 m between rows and 0.2 m between plants. The three maize lines were planted randomly in each plot. Plants were harvested at time points during the transition from the vegetative to the reproductive stage of growth (30 days), at flowering (60 days) and during grain filling and maturity (90 days). At each time point, three sets of three plants of each cultivar were harvested, after which the roots were shaken manually to remove the loosely-attached soil. The soil adhering to the roots of each set of three plants was then pooled, homogenized and considered as the rhizosphere soil. This yielded three replicates per treatment. The samples were kept at −20 °C until DNA extraction and further processing.
Extraction of DNA from rhizosphere soil samples
Total microbial community DNA was extracted directly from the rhizosphere soil samples (0.5 g of each sample in triplicate) using the FastDNA® Spin Kit For Soil (BIO 101 Systems, USA). DNA preparations were visualized by electrophoresis in a 0.8 % agarose gel in 1× TBE buffer (Sambrook et al. 1989) to assess yield, integrity and average fragment size and then stored at 4 °C prior to PCR amplification.
PCR amplifications of 16S rRNA gene for total bacteria, actinobacteria and internal transcribed spacer (ITS) regions of fungi for DGGE analysis
The amplification of 16S ribosomal RNA gene fragments (rRNA) was performed using 50 μl reactions containing 10 ng of genomic DNA and 0.2 mM of 16S rRNA gene universal primers 968F-GC (5′-GC clamp- AACGCGAAGAACCTTAC3′) and R1401 (5′ CGGTGTGTACAAGACCC3′), 2.5 mM MgCl+2, 0.2 mM dNTPs, 2U Taq polymerase in the buffer supplied by the manufacturer (Promega, USA). The amplification conditions were 30 cycles of 1 min 94 °C; 1 min 55 °C; 2 min 72 °C. A hot start of 2 min 94 °C was applied to avoid initial mispriming and enhance the specificity. A final extension step (10 min 72 °C) was run and the reaction tubes were then cooled to 4 °C (Muyzer et al. 1993; Nübel et al. 1996). Amplifications of 16S rRNA gene fragments of Actinobacteria were performed using a semi-nested protocol. The first PCR reaction was performed using 25 μl reactions containing 10 ng of genomic DNA and 0.1 mM specific primer for Actinobacteria denoted 243F (5′GGA TGA GCC CGC GGC CTA3′), in combination with primer 1401R, 0.2 mM MgCl+2, 0.2 mM dNTPs, 5 U Taq DNA polymerase and the buffer supplied by the manufacturer (Promega). Amplification conditions were as follows: initial denaturation for 5 min at 94 °C; 35 cycles of 1 min at 94 °C, 1 min at 58 °C, and 2 min at 72 °C; a final extension for 10 min at 72 °C; cooling to 4 °C. (Nübel et al. 1996; Heuer et al. 1997). Amplicons obtained in this first PCR reaction were then used as templates for a second amplification with primers 968F-GC and R1401 (Nübel et al. 1996). The reaction mixture was the same as described above.
Fungal amplicons based on the ITS region were obtained using a nested protocol. The first PCR reaction was performed using 25 μl reactions containing 10 ng of genome DNA and 0.4 mM of the primers EF4F (5′GGAAGGGRTGTATTTATTAG3′) and ITS4R (5′TCCTCCGCTTATTGATATGC3′), 0.2 mM dNTPs, 2 mM MgCl+2, 10 U Taq DNA polymerase in the buffer supplied by the manufacturer (Promega, USA). The amplification conditions were a initial denaturation step for 5 min at 94 °C; 34 cycles of 30 s at 94 °C, 30 s at 55 °C, 1 min 30 s at 72 °C; a final extension for 5 min at 72 °C; cooling to 4 °C. Amplicons obtained in this first PCR reaction were then used as template for a second amplification procedure using 0.4 mM of the primers ITS1F-GC (5′-GC clamp—CTTGGTCATTTAGAGGAAGTAA3′) and ITS2R (5′GCTGCGTTCTTCATCGATGC3′), 0.25 mM dNTPs, 2 mM MgCl+2, 5 U Taq DNA polymerase in buffer supplied by the manufacturer, with total volume of reaction mix of 50 μl. Amplification conditions were as described by Anderson et al. (2003a, b): initial denaturation for 5 min at 94 °C; 34 cycles of 30 s at 94 °C, 30 s at 55 °C, 30 s at 72 °C; a final extension for 5 min at 72 °C; cooling to 4 °C.
DGGE and statistical analyses
DGGE analysis was carried out as described previously (Muyzer et al. 1993) using the Ingeny phorU2 apparatus (Ingeny International, The Netherlands). Thirty μl of PCR products were loaded onto 8 % (w/v) polyacrylamide gels in 1× TAE buffer (20 mM Tris–acetate, pH 7.4, 10 mM acetate, 0.5 mM disodium EDTA). Polyacrylamide gels contained a denaturing gradient of urea and formamide varying from 45 to 65 % and from 20 to 55 % for total bacteria/actinobacteria and fungi, respectively. The gels were run for 16 h at 100 V and 65 °C. After this period, they were stained for 1 h in SYBR Green I nucleic acid staining solution (10× concentrated) (Molecular Probes, The Netherlands) and immediately photographed under UV light. Dendrograms were constructed based on the presence and absence of bands with the unweighted pair group method (UPGMA) with mathematical averages and similarity coefficient of Pearson using the BioNumerics software (Applied Maths, Ghent, Belgium). The gels were analyzed to check for heterogeneity of the data set through detrended correspondence analysis (DCA). Data sets with gradient lengths shorter than 3.0 were considered to be homogeneous, while those with gradient lengths longer than 4.0 were considered to be heterogeneous (Leps and Smilauer 2003). To evaluate the distribution of the samples, principal components analyses (PCA) were performed using the CANOCO statistical software (CANOCO 4.55, Biometris, Wageningen, Netherlands) as described previously (Leps and Smilauer 2003). To correlate the profile obtained with environmental variables (such as soil type and period of cultivation), redundancy analysis (RDA) was used. In order to investigate the significance of the relations, Monte Carlo permutation tests based on 499 random permutations were performed, taking as the null hypothesis that species data were unrelated to environmental data and as the alternative hypothesis that the species responded to the environment (Leps and Smilauer 2003; Salles et al. 2004). To approximate bacterial, fungal and actinobacterial richnesses, we used band (species) richness S, derived from the number of DGGE-detectable bands. It is important to note that this provided just a crude proxy for richness, which was used here for comparative purposes. Similarly, evenness was roughly estimated on the basis of the relative band intensities. Together, this yielded the Shannon-Wiener index of bacterial, fungal and actinobacterial diversity, H, which crudely describes the diversity of the dominant species in soil microbial community (Gelsomino et al. 1999). The index was calculated as H = ∑Pi log Pi, based on the relative band intensities (Pi) as formulated by Eichner and co-workers (Eichner et al. 1999). Means and standard deviations of S and H were calculated for every treatment. Test t were used to distinguish significant differences in H for bacterial/actinobacterial and fungal communities (Jurelevicius et al. 2010; Semenov et al. 2012).
Quantitative real-time PCR analysis of bacteria
Bacterial abundances were quantified by real-time PCR (qPCR) using primers PF16S and PR16S (Bach et al. 2002). The qPCR reactions were performed using an ABI prism 7300 Cycler (Applied Biosystems, Germany) and a SYBR Green I fluorophore protocol as recommended by the manufacturer (Applied Biosystems). Amplification was carried out in 25-μl reaction volumes containing 2 μl of diluted template DNA, 40 μM of each primer, 7.75 μl of PCR-grade water, 1.25 μl of bovine serum albumin (BSA) and 12.5 μl of LightCycler Faststartplus DNA Master SYBR Green I (Roche, Germany). All amplifications were performed in triplicate. PCR-grade water (no template) was used as a negative control throughout. Thermal cycling for the assays consisted of 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 20 s, annealing at 62 °C for 1 min, and elongation at 72 °C for 1 min. Fluorescence signals were measured once per cycle at the end of the denaturation step. After amplification, a melting curve was obtained by heating the products to 95 °C, cooling them to 65 °C and then gradually heating them to 95 °C at a rate of 0.2 °C per second. The specificity of the qPCR products was confirmed by melting curve analysis and gel-based post-PCR analysis. Triplicate results of real-time PCR measurements were averaged and the standard error was calculated. Standard curves were constructed based on triplicate 10-fold dilutions of a DNA standard containing a known number of target gene copies of Burkholderia terrae strain BS001.
Results
Plant growth and sample characteristics
All maize genotypes (non-Bt, MON810 and TC1507) revealed similar growth rates during the “Safra” and “Safrinha” crop seasons and in both soil types (Cerrado and Várzea). No signs of disease or nutrient scarcity were noted (data not shown). Thus, sampling of the respective rhizospheres was performed as described.
Composition of bacterial, actinobacterial and fungal communities in the maize rhizosphere
The bacterial, actinobacterial and fungal communities were evaluated with respect to their structures in the rhizosphere soil samples. Per soil, time and genotype, highly reproducible PCR-DGGE profiles were obtained between the triplicate rhizosphere soil samples from all communities evaluated in our experiment, indicating the robustness of the PCR-DGGE analyses (data not shown). Due to the high sample numbers and to avoid the gel-to-gel bias, one DGGE gel was performed with one replicate of each sample (genotype, soil and time) for each evaluated community. Overall, the PCR-DGGE patterns were complex across the rhizospheres of the Bt and non-Bt maize plants in both soils (Fig. 1). Across the board, the bacterial (Fig. 1a, b), actinobacterial (Fig. 1c, d) and fungal (Fig. 1e, f) profiles differed between soils, indicating soil type was an important factor shaping these communities. The profiles also revealed shifts in the community structures between “Safra” and “Safrinha” seasons, and to a lesser extent at different plant developmental stages within these periods, in the same soil, which indicated time/plant growth period was another effector of the community structures (Fig. 1). Maize genotype apparently did not affect the community structures to a significant extent, although the clustering was quite erratic among the different samples. The cluster analysis revealed that the non-Bt- and Bt-maize TC1507 samples (90 days) grouped together and were separated from Bt-maize MON810 (Fig. 1a) at a high level (95 %) of similarity. This suggested a great similarity among the three communities, in spite of the clustering in groups. Possibly, the microbial communities were not strongly driven by the GM maize genotypes, independently of the evaluated community, soil type and period of maize cultivation (Fig. 1).

DGGE fingerprint analyses of total bacterial, actinobacterial and fungal communities in both Cerrado and Várzea soils. (a) bacteria, (c) actinobacteria and (e) fungi are from Cerrado soil samples while (b) bacteria, (d) actinobacteria and (f) fungi from Várzea soil samples. The transgenic Bt-maize lines are represented by transformation event MON810 and transformation event TC1507. The isogenic non-transgenic line is represented by the non-Bt maize genotype. The period of maize cultivation in Brazil is represented by the number 2 (“Safrinha”) and the absence of this number corresponds to the “Safra” period. The maize growth stages (when they were harvested for sampling) are represented by 30 (transition from vegetative to reproductive stage of growth), 60 (flowering) and 90 (grain filling and maturity)
Ordination of the PCR-DGGE profiles using PCA supported the aforementioned effects of soil and time/plant developmental stage on the bacterial, actinobacterial and, to a lesser extent, the fungal communities. The first PCA axes explained 22.6, 25.0 and 26.4 % of the variance, whereas the second ones covered 21.0, 22.1 and 13.1 % (Fig. 2a–c). With respect to the total bacterial communities, there was a tendency for these communities to group in accordance with soil type along the first and second axes, but it was not possible to clearly separate the samples in function of maize genotype and/or growth stage of the crop (Fig. 2a). Along the second axis, the samples were loosely grouped in function of soil type, given that samples of Cerrado soil/“Safra” occurred primarily in the upper part and samples of Várzea soil/“Safrinha” in the lower part of the graph. Specifically, the PCA separated, along the first axis, the actinobacterial communities from “Safra” and “Safrinha” (Fig. 2c) whereas, along the second axis, separation occurred in accordance with soil type (Fig. 2c). Furthermore, the PCA of the fungal PCR-DGGE profiles separated, along the first axis, the samples from the Cerrado and Várzea soils (Fig. 2b). Along the second axis, separation occurred between the “Safra” and “Safrinha” periods, primarily for the Várzea soil (Fig. 2b).

Principal component analyses (PCA) was made from DGGE patterns of the bacterial (a), fungal (b) and actinobacteria (c) genes. Values of the first (22.6, 26.4 and 25.0 %) and second axes (21.0, 13.1 and 22.1 %) indicate the variance explained by each dimension. The soils were represented by letter c for Cerrado soil and letter v for Várzea soil. The transgenic Bt-maize line transformation event MON810 is represented by the letter y and the transformation event TC1507 by the letter h. The isogenic non-transgenic line was represented by the absence of a letter. The period of maize cultivation in Brazil is represented by the number 2 (“Safrinha”) and the absence of this number corresponds to the “Safra” period. The maize growth stages (when they were harvested for sampling) are represented by 30 (transition from vegetative to reproductive stage of growth), 60 (flowering) and 90 (grain filling and maturity)
To estimate the species distribution through an overview of the compositional gradients of the data, the PCR-DGGE patterns were converted into matrices and subjected to detrended correspondence analysis (DCA). The DCA showed a linear distribution of the data, indicating homogeneity, with gradient lengths for the first axis of 2.643, 2.374 and 2.343 for bacteria, Actinobacteria and fungi, respectively. We thus inferred that RDA provided a proper mathematical model for multivariate analysis to make inferences about the environmental variables (type of soil, period of crop and plant genotype) that affect the structures of the bacterial, actinobacterial and fungal communities (Table 1). The RDA analysis of the data set revealed that the samples indeed grouped in function of soil type, irrespective of the plant line evaluated (Table 1). There was no difference in the bacterial communities between the non-Bt and the two Bt maize lines (p > 0.05). A Monte Carlo permutation test indicated that soil type was the main factor determining the composition of the bacterial communities in the rhizosphere of the maize plants (p = 0.002). Therefore, the period of cultivation and plant growth were not significant as explanatory variables (p > 0.05). The actinobacterial and fungal communities showed the same profile assembly, with samples grouped in function of soil type and crop period independently of maize genotype (p = 0.002; p = 0.002).
Diversity of bacteria and fungi as affected by the GM maize
On the basis of the PCR-DGGE data, the Shannon indices of diversity of bacterial, actinobacterial and fungal communities were estimated. The index such as used herein has to be interpreted with caution within the known limitations of the soil DNA based PCR-DGGE approach. In relation to the bacterial communities, the Shannon index values were higher in the Várzea than in the Cerrado soil (Fig. 3a). Fluctuations in this index, in function of plant age and period of crop, can be observed. In both soils, similar patterns of fluctuation were found, without any variation related to plant genotype. Specifically, a decrease of the index value in non-Bt maize during the “Safra” period was found between 30 and 90 days, with values ranging from 2.44 ± 0.02 to 1.10 ± 0.02. In the “Safrinha” period, we did not see a similar trend in the profile, probably related to different rate of rain and temperature. The MON810 line showed a decreasing H value from 30 to 60 days followed by a small increase after 90 days, with values ranging from 1.94 ± 0.01 to 1.77 ± 0.02, with the same profile being observed in “Safrinha”. In Várzea soil, the samples showed the same trends observed in Cerrado soil, with fluctuations in relation to plant age (Fig. 3a). As in total bacteria, the index for actinobacteria showed clear differences between the soil types. A slight variation was related to plant age, but again no differences were observed between the different maize lines. A small decrease in the index values was observed with plant age in “Safra” and “Safrinha”. During “Safra” in both soils, it was not possible to observe any fluctuation related to plant genotype (Fig. 3b). Concerning the fungal communities, we observed a fluctuation as a function of plant age and crop period without relation with plant genotype, during the two periods of cultivation and across soils (Fig. 3c). In both communities evaluated it was not possible to show any significant changes (p > 0.05).

Changes in bacterial (a), actinobacterial (b) and fungal (c) diversity (Shannon index, H) for non-Bt and Bt-maize cultivated during two different periods of the year in two different soils. The transgenic Bt-maize lines are represented by transformation event MON810 and transformation event TC1507. The isogenic non-transgenic line is represented by the non-Bt maize genotype. The period of maize cultivation in Brazil is represented by the number 2 (“Safrinha”) and the absence of this number corresponds to the “Safra” period. The maize growth stages (when they were harvested for sampling) are represented by 30 (transition from vegetative to reproductive stage of growth), 60 (flowering) and 90 (grain filling and maturity). The error bars indicate standard error
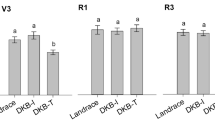
Abundance of 16S ribosomal RNA (rRNA) genes in the maize rhizospheres
Ribosomal RNA gene copy numbers were quantified using quantitative PCR on the basis of the 16S rRNA genes. The size of the amplified regions was 279 bp, amplification efficiencies were 99.0 % and R 2 values averaged 0.99. The detection limit was 100 copies of the target region per reaction. The data showed that it was not possible to observe any differences in the 16S rRNA gene copy numbers between the Bt- and non-Bt maize lines, regardless of soil type and period of crop (Fig. 4). Per soil, the 16S rRNA gene abundances were in the range 9.55 × 109 to 9.95 × 109 g−1 for Cerrado soil and 9.33 × 109 to 10.01 × 1010 g−1 for Várzea soil. For the non-Bt maize cultivated in both soils, we observed no differences in the abundance of the 16S rRNA genes between two periods of crop. In relation to Cerrado soil, slight differences were observed in relation to the stage of plant growth. This was probably caused by the selection of specific organisms in the rhizosphere related to the most active period of plant root exudation, in which an increase during plant maturation was observed. At 30 days post-germination, the 16S rRNA gene copy numbers per g rhizosphere soil were 9.27 × 109, increasing to 9.55 × 109 at 60 days and 9.75 × 109 at 90 days. The TC1507 line showed the same bacterial abundance as that observed in the non-Bt maize while the MON810 genotype did not show any differences between the two periods of crop and between the stages of plant growth (Fig. 4). It was not possible to observe a trend in the abundance data for the Várzea soil. In relation to the first crop, the TC1507 (9.92 × 109 to 9.80 × 109) genotype showed high values of abundance in relation to the non-Bt and MON810 genotype (9.34 × 109 to 9.78 × 109). The MON810 genotype did not show any differences between plant growth and period of crop.

Abundance of bacterial 16S rRNA coding gene in the rhizospheres of different maize cultivated in two different periods of the year in two soil types. The error bars indicate standard error
Discussion
In soil, a wide range of factors affect microbial life including plant type, soil type, agricultural management regime and the interaction between these factors (van Veen et al. 1997; Garbeva et al. 2004). Plant type may influence the composition and structure of the root-associated microbial communities mainly via root exudation of sugars, organic acids, amino acids, proteins and other compounds. Such compounds can be used by the local microorganisms as energy and carbon sources. Each plant type may show its own typical exudation pattern and microorganisms respond differently to the compounds released by the plant root. Different root exudates are expected to select different rhizosphere communities (Garbeva et al. 2004). The GM plants studied here had functional Bt genes inserted in the genomes, which are known to be constitutively expressed in all stages of plant growth (Saxena et al. 2002; Icoz and Stotzky 2007). The Bt-protein produced by these genes was likely secreted into the plant root exudates and thus could affect the local soil microbiota. This process might therefore select microorganisms that can use the Bt protein as a carbon source (di Giovanni et al. 1999; Baumgarten and Tebbe 2005). Moreover, an additional event that should affect microbial communities in soil is related with the possibility of bacterial rhizosphere populations to capture and stably integrate transgenic plant DNA, in particular antibiotic resistance genes used as markers in transgenic crops (Lynch et al. 2004). These events, in particular the former one, can influence specific microbial populations, resulting in a modification of the soil functionality (Lamarche and Hamelin 2007). However, the extent to which such events take place is still a controversial issue (Dively et al. 2004; Baumgarte and Tebbe Baumgarten and Tebbe 2005; Castaldini et al. 2005).
We thus evaluated the possible effects of two Bt maize plants cultivated in two different tropical soil types during two different periods of cultivation and during different plant growth stages using molecular techniques. Remarkably, we did not find any conspicuous differences between the two GM lines and the non-transgenic line in relation to the diversities of evaluated microbial communities. We did, however, observe shifts in the composition of microbial communities and could relate these to the factors of soil type, plant age and period of cultivation.
It is known that soil type is an important factor that determines soil microbial community structure in agricultural systems, the main reason being that soil determines the habitable niches in accordance with nutrient availability and local conditions (Garbeva et al. 2004). In the case of the Bt-maize, soil is a key parameter that likely governs the exposure to Bt-toxin of soil organisms. For instance, the protein could be retained in soil particles, encompassed with clay, for long periods of time (Saxena et al. 2002, Badea et al. 2010). We selected two different soil types based on their usage for maize production. Interestingly, differences in the structures of all three communities that were evaluated in function of soil type were clearly observed. Our data corroborate those observed in a previous study (Seldin et al. 1998). Other studies have also demonstrated that soil type and texture, rather than maize genotype, were the overriding factors that influenced the root-associated microbial community structures (Chiarini et al. 1998; da Silva et al. 2003). Strikingly, no difference in the function (growth) of the maize lines evaluated was observed, the most likely reason for this being the composition of the soil that allowed growth. Moreover, in soils with high clay content, the Cry protein might adsorb on the clay-containing structures, becoming unavailable for microbial utilisation. Thus, considering the high clay contents of both our soils, we may infer that an adsorption phenomenon may have caused the absence of any change in the structure of the three communities that were evaluated. This is in agreement with previous studies (Blackwood and Buyer 2004; Badea et al. 2010; Helassa et al. 2011).
Other important factors that correlated with the shift in the community structures that were measured were plant age and cropping season. We observed differences in the structures of all three microbial communities evaluated in relation to cropping season, much as described recently (Chun et al. 2011). In our study, the maize cultivation periods differed remarkably, with different amounts of rain (300 mm for the rainy period and 60 mm for the dry period) and variations of temperature. It is likely that certain groups of soil microbes are adapted to particular soil temperature and moisture regimes. Fluctuations in soil temperature and humidity stimulate the growth of specific groups of soil microbes, causing variations or shifted community structures (Chun et al. 2011). Factors like plant age and field heterogeneity may be stronger drivers of community structures than the insertion of an exogenous gene in the plant (Baumgarten and Tebbe 2005). We also did not observe any variation in the abundance of total bacteria as a function of plant genotype. In spite of the finding of a minor difference in relation to plant growth stage, type of crop and soil, the data were very consistent and indicated a high and rather constant carrying capacity for bacteria in the maize rhizospheres in the two soils. Similar results have been recently described in a study on transgenic rice, in which total bacteria and fungi were evaluated (Lee et al. 2011). Donegan et al. (1999) found higher densities of bacteria in the rhizosphere of Bt plant lines than in those of conventional lines, in contrast to Rui et al. (2005), who found higher numbers in conventional plants. Another study (Saxena and Stotzky 2001) did not observe any differences in the communities between transgenic and non-transgenic plants. In these studies, the authors did not directly correlate the release of the Bt-toxin with an increase or decrease in bacterial numbers, indicating that other factors are involved in these shifts. Icoz and Stotzky (2008) described that the impact of genetic modification of crop plants on the composition of the soil microbial communities is generally transient and minor compared with the natural variations that are a function of growing season, field site and soil type. The same minor impact has been observed in a long-term study (four years) of cultivation of transgenic maize (Barriuso et al. 2012).
We conclude that the effects of two Bt maize lines growing in two typical tropical soils on the root-associated bacterial, Actinobacterial and fungal communities are likely to be very minor when compared to those of the near-isogenic parental line. In the well-replicated field studies reported here, no evidence of such effects on the above mentioned communities were discerned. In contrast, expected effects of soil type, plant growth stage and season of growth were clearly detectable. The absence of major effects of the Cry-gene-loaded GM maize plants on the soil microbiota provides another piece of evidence for the lack of impact on soil of GM crop plants.
References
Anderson IC, Campbell CD, Prosser JI (2003a) Diversity of fungi in organic soils under a moorland-Scots pine (Pinus sylvestris L.) gradient. Environ Microbiol 11:1121–1132
Anderson IC, Campbell CD, Prosser JI (2003b) Potential bias of fungal 18S rRNA and internal transcribed spacer polymerase chain reaction primers for estimating fungal biodiversity in soil. Environ Microbiol 1:36–47
Andreote FD, Mendes R, Dini-Andreote F, Rossetto PB, Labate CA, Pizzirani-Kleiner AA, van Elsas JD, Azevedo JL, Araújo WL (2008) Transgenic tobacco revealing altered bacterial diversity in the rhizosphere during early plant development. Antonie Van Leeuwenhoek 93:415–424
Andreote FD, Carneiro RT, Salles JF, Marcon J, Labate CA, Azevedo JL, Araújo WL (2009) Culture-independent assessment of Alphaproteobacteria related to order Rhizobiales and the diversity of cultivated Methylobacterium in the rhizosphere and rhizoplane of transgenic eucalyptus. Microb Ecol 57:82–93
Bach HJ, Tomanova J, Schloter JC, Munch JC (2002) Enumeration of total bacteria with genes for proteolytic activity in pure cultures and in environmental samples by quantitative PCR mediated amplification. J Microbiol Methods 49:235–245
Badea EM, Chelu F, Lacatusu A (2010) Results regarding the levels of Cry1Ab protein in transgenic corn tissue (MON810) and the fate of Bt protein in three soil types. Rom Biotechnol Lett 15:55–62
Barriuso J, Valverde JR, Mellado RP (2012) Effect of Cry1Ab protein on rhizobacterial communities of Bt-maize over a four-year cultivation period. PLoS ONE 7:e35481
Baumgarten S, Tebbe CC (2005) Field studies on the environmental fate of the Cry1Ab Bt-toxin produced by transgenic maize (MON810) and its effect on bacterial communities in the maize rhizosphere. Mol Ecol 14:2539–2551
Blackwood CB, Buyer JS (2004) Soil microbial communities associated with Bt and non-Bt corn in three soils. J Environ Qual 33:832–836
Bruinsma M, Kowalchuk GA, van Veen JA (2003) Effects of genetically modified plants on microbial communities and processes in soil. Biol Fertil Soils 37:329–337
Carpenter JE (2011) Impacts of GM crops on biodiversity. GM Crops 1:1–17
Castaldini M, Turrini A, Sbrana C, Beneditti A, Marchionni M, Mocali S, Fabiani A, Landi S, Santomassimo F, Pietrangeli B, Nuti MP, Miclaus N, Giovannetti M (2005) Impact of Bt corn in rhizospheric and soil eubacterial communities and on beneficial mycorrhizal symbiosis in experimental microcosms. Appl Environ Microbiol 71:6719–6729
Chauhan PS, Chaudhry V, Mishra S, Nautiyal CS (2011) Uncultured bacterial diversity in tropical maize (Zea mays L.) rhizosphere. J Basic Microbiol 51:15–32
Chiarini L, Bevivino A, Dalmastri C, Nacamulli C, Tabacchioni S (1998) Influence of plants development, cultivar and soil type on microbial colonization of maize root. Appl Soil Ecol 8:11–18
Chun YJ, Kim DY, Kim HJ, Park KW, Jeong S-C, Park S, Lee B, Ham CH, Kim HM, Kim C-G (2011) Do transgenic chili pepper plants producing viral coat protein affect the structure of a soil microbial community? Appl Soil Ecol 51:130–138
da Silva KRA, Salles JF, Seldin L, van Elsas JD (2003) Application of a novel Paenibacillus-specific PCR-DGGE method and sequence analysis to assess the diversity of Paenibacillus spp. in the maize rhizosphere. J Microbiol Methods 54:213–231
di Giovanni GD, Watrud LS, Seidler RJ, Widmer F (1999) Comparison of parental and transgenic alfalfa rhizosphere bacterial communities using BIOLOG GN metabolic fingerprinting and Enterobacterial Intergenic Consensus Sequence-PCR (ERIC-PCR). Microbiol Ecol 37:129–139
Dively GP, Rose R, Sears MK, Hellmich RL, Stanley-Horn DE, Calvin DD, Russo JM, Anderson PL (2004) Effects on monarch butterfly larvae (Lepidoptera: Danaidae) after continuous exposure to Cry1Ab-expressing corn during anthesis. Ann Entomol Soc Am 33:1116–1125
Donegan KK, Seidler RJ, Porteous LA, Digiovanni G, Widmer F, Watrud LS (1999) A field study with genetically engineered alfafa inoculated with recombinant Sinorhizobium meliloti: effects on the soil ecosystem. J Appl Ecol 6:920–936
Dunfield KE, Germida JJ (2004) Impact of genetically modified crops on soil-and plant-associated microbial communities. J Environ Qual 33:806–815
Eichner CA, Erb RW, Timmis KN, Wagner-Dobler I (1999) Thermal gradient gel electrophoresis analysis of bioprotection from pollutant shocks in the activated sludge microbial community. Appl Environ Microbiol 65:102–109
Feng Y, Ling L, Fan H, Liu Y, Tan F, Shu Y, Wang J (2011) Effects of temperature, water content and pH on degradation of Cry1Ab protein released from Bt corn straw in soil. Soil Biol Biochem 43:1600–1606
Garbeva P, van Veen JA, van Elsas JD (2004) Microbial diversity in soil: selection of microbial populations by plant and soil type and applications for disease suppressiveness. Annu Rev Phytopathol 42:243–270
Gelsomino A, Keijzer-Wolters AC, Cacco G, van Elsas JD (1999) Assessment of bacterial community structure in soil by polymerase chain reaction and denaturing gradient gel electrophoresis. J Microbiol Methods 38:1–15
Gruber H, Paul V, Meyer HHD, Müller M (2012) Determination of insecticidal Cry1Ab protein in soil collected in the final growing seasons of a nine-year field trial of Bt-maize MON810. Transgenic Res 21:77–88
Hanulla SE, Boschker HTS, de Boer W, van Veen JA (2012) 13C pulse-labeling assessment of the community structure of active fungi in the rhizosphere of a genetically starch-modified potato (Solanum tuberosum) cultivar and its parental isoline. New Phytol 194:784–799
Helassa N, Charek AM, Quiquampoix H, Noinville S, Déjardin P, Frutos R, Staunton S (2011) Effects of physicochemical interactions and microbial activity on the persistence of Cry1Ab Bt (Bacillus thuringiensis) toxin in soil. Soil Biol Biochem 5:1089–1097
Heuer H, Krsek M, Baker P, Smalla K, Wellington EMH (1997) Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel electrophoretic separation in denaturing gels. Appl Environ Microbiol 8:3233–3241
Icoz I, Stotzky G (2007) Cry3Bb1 protein from Bacillus thuringiensis in root exudates and biomass of transgenic corn does not persist in soil. Transgenic Res 4:609–620
Icoz I, Stotzky G (2008) Fate and effects of insect-resistant Bt crops in soil ecosystems. Soil Biol Biochem 40:559–586
James C (2010) Global status of commercialized biotech/GM Crops: ISAAA Brief No. 42. ISAAA: Ithaca, NY
Jurelevicius D, Korenblum E, Casella R, Vital RL, Seldin L (2010) Polyphasic analysis of bacterial community in the rhizosphere and roots of Cyperus rotundus L. grown in a petroleum-contaminated soil. J Microbiol Biotechnol 5:862–870
Lamarche J, Hamelin RC (2007) No evidence of an impact on the rhizosphere diazotroph community by the expression of Bacillus thuringiensis Cry1Ab toxin by Bt white spruce. Applied Environ Microbiol 73:6577–6583
Lee S-H, Kim C-G, Kang H (2011) Temporal dynamics of bacterial and fungal communities in a genetically modified (GM) rice ecosystem. Microbiol Ecol 61:646–659
Leps J, Smilauer P (2003) Multivariate analysis of ecological data using CANOCO. University Press, Cambridge
Lynch JM, Benedetti A, Insam H, Nuti MP, Smalla K, Torsvik V, Nannipieri P (2004) Microbial diversity in soil: ecological theories, the contribution of molecular techniques and the impact of transgenic plants and transgenic microorganism. Biol Fertil Soils 40:363–385
McLaren J, Copping L (2011) Transgenic maize—the registration status of lines that have been commercialised: the first in a series that examines the GM crop market. Outlooks Pest Manag 2:66–73
Motavalli PP, Kremer RJ, Fang M, Means NE (2004) Impact of genetically modified crops and their management on soil microbial mediated plant nutrient transformations. J Environ Qual 33:816–824
Muyzer G, de Wall EC, Uitterlinden AG (1993) Profiling of complex microbial population by denaturing gradient gel electrophoresis analysis of polymerase chain reaction—amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Nübel U, Engelen B, Felske A, Snaidr J, Wieshuber A, Amann RI, Ludwig W, Backhaus H (1996) Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J Bacteriol 19:5636–5643
Raybould A, Higgins LS, Horak MJ, Layton RJ, Storer NP, de la Fuente JM, Herman RA (2011) Assessing the ecological risks from the persistence and spread of feral populations of insect-resistant transgenic maize. Transgenic Res 3:55–64
Rui YK, Yi GX, Zhao J, Wang BM, Li ZH, Zhai ZX, He ZP, Li QX (2005) Changes of Bt toxin in the rhizosphere of transgenic Bt cotton and its influence on soil functional bacteria. World J Microbiol Biotechnol 21:1279–1284
Salles JF, van Veen JA, van Elsas JD (2004) Multivariate analyses of Burkholderia species in soil: effect of crop and land use history. Appl Environ Microbiol 70:4012–4020
Sambrook J, Fritsch EF, Manniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Saxena D, Stotzky G (2001) Bacillus thuringiensis (Bt) toxin released from root exudates and biomass of Bt corn has no apparent effect on earthworms, nematodes, protozoa, bacteria, and fungi in soil. Soil Biol Biochem 33:1225–1230
Saxena D, Flores S, Stotzky G (2002) Bt toxin is released in root exudates from 12 transgenic corn hybrids representing three transformation events. Soil Biol Biochem 34:133–137
Seldin L, Rosado AS, da Cruz DW, Nobrega A, van Elsas JD (1998) Comparison of Paenibacillus azotofixans isolated from rhizoplane, rhizosphere, and non-root associated soil from maize planted in two different Brazilian soils. Appl Environ Microbiol 64:3860–3868
Semenov AV, Pereira e Silva MC, Szuturc-Koestsier AE, Schmitt H, Salles JF, van Elsas JD (2012) Impact of incorporated fresh 13C potato tissues on the bacterial and fungal community composition of soil. Soil Biol Biochem 49:88–95
Tan F, Wang J, Feng Y, Chi G, Kong H, Qiu H, Wei S (2010) Bt corn plants and their straw have no apparent impact on soil microbial communities. Plant Soil 329:349–364
Tan F, Wang J, Chen Z, Feng Y, Chi G, Rehman SU (2011) Assessment of the arbuscular mycorrhizal fungal community in roots and rhizosphere soils of Bt corn and their non-Bt isolines. Soil Biol Biochem 43:2473–2479
Van Veen JA, van Overbeek LS, van Elsas JD (1997) Fate and activity of microorganisms introduced into soil. Microbiol Mol Biol Rev 61:121–135
Yu HL, Li YH, Wu KM (2011) Risk assessment and ecological effects of transgenic Bacillus thuringiensis crops on non-target organisms. J Integrative Plant Biol 7:520–538
Acknowledgments
This study was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ). Thanks are also due to the Conselho Nacional de Pesquisa (CNPq) for providing a fellowship to Simone Cotta to come over to the Groningen Microbial Ecology laboratory. The Soil Biotechnology Foundation is acknowledged for providing financial support to Armando C F Dias and received a graduate fellowship from FAPESP (no. 2008/54013-8).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Cotta, S.R., Dias, A.C.F., Marriel, I.E. et al. Temporal dynamics of microbial communities in the rhizosphere of two genetically modified (GM) maize hybrids in tropical agrosystems. Antonie van Leeuwenhoek 103, 589–601 (2013). https://doi.org/10.1007/s10482-012-9843-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-012-9843-7