
Abstract
Polymerase chain reaction–denaturing gradient gel electrophoresis (PCR–DGGE) and sequences of the 16S and 18S rRNA genes were used to access the effects of actively growing Bacillus thuringiensis (Bt) corn Pioneer 34B24 and Nongda 1246*1482, and plant straw (leaves plus stalks) of Bt hybrid Pioneer 34B24 and Nongda 61 on soil bacterial and fungal communities. Two-way indicator species analysis (TWINSPAN®), and detrended correspondence analysis (DCA) of the DGGE data indicated that neither the actively growing Bt corn nor its straw had any constant apparent effect on the soil bacteria and fungi community structure. The age of the growing plants, or the timing of plant straw decomposition may have more effect on the microbial community than other factors, i.e., the presence of Cry protein, plant hybrid and variety.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Genetically modified (GM) crop plants for resistance to pests represent a potential promising tool with which to decrease the amount of chemical pesticides used in agriculture. Cry-proteins from Bacillus thuringiensis (Bt) are by far the most common insecticidal proteins that have been engineered into plants. Bt cry genes have been engineered into a large number of plant species, such as corn, cotton, potato, tomato, eggplant, rice, etc. (see, e.g., Icoz et al. 2008; Sanvido et al. 2007). Bt corn is grown on about 11.3 million ha worldwide and is, by far, the most widely grown Bt crop in the world (James 2005). The “first generation” Cry proteins engineered into corn were Cry1Ab, and Cry9C. Monsanto’s YieldGard® corn, transformation event MON810, expressing Cry1Ab toxic to some lepidopteran insect pests, is the most widely grown Bt crop today (Benedict and Ring 2004; Icoz and Stotzky 2008).
In China, Bt corn has not yet been approved for commercial cultivation, although evaluation of the environmental risk of some released Bt corn hybrids [e.g., Pioneer 34B24 (Mon810) from Monsanto, expressing Cry1Ab toxin, and Nongda 61 and Nongda 1246*1482 from China Agricultural University, expressing Cry1A protein] in the North East Plain and North China Plain has lasted for more than 10 years (1998–2008). The aim of introducing Bt corn to China is to control the damage of Asian corn borer Ostrinia furnacalis (Guenee) and reduce pesticide input in corn production. The delayed commercial introduction of Bt corn in China results mainly from the following: firstly, cultivating Bt corn may lead to development of resistance of the target pests (i.e., Ostrinia furnacalis [Guenee] and Helicoverpa armigera [Hubner]) of Bt cotton. The North East and North China Plains are the main cultivated Bt cotton belt, as well as being the most suitable region for corn cultivation in China. By 2006, Bt cotton had been grown intensively in every cotton-growing area, and had reached 4 million ha, accounting for 70% of the total growing area (Wu 2007). The conventional corn fields in these regions had provided a natural refuge for management of the resistance to Bt cotton by H. armigera (Hubner). Secondly, the release of Bt corn remains a long-term concern because of the presumed potential ecological and environmental risks, e.g., effects on non-target organisms, effects on soil ecosystems, etc. (Icoz et al. 2008; Saxena et al. 2004; Sanvido et al. 2007).
To date, laboratory and field studies have demonstrated that Cry protein expressed in Bt corn enters the soil system mainly via root exudation throughout the growth of the plant, in pollen released during tasseling, and from crop residues after harvest (e.g., Icoz and Stotzky 2007; Icoz et al. 2008; Lehman et al. 2008; Saxena et al. 1999, 2002a; Saxena and Stotzky 2000; Zscheischler et al. 1984; Zwahlen et al. 2003). As a toxin, Cry protein released to soil from Bt corn has been shown to degrade slowly and to accumulate in soil (Tapp and Stotzky 1998; Saxena et al. 2002a; Zwahlen et al. 2003). Organisms in soil will come into contact with transgenic Cry proteins when the proteins are released from Bt corn in root exudates or from decomposing plant tissue, thus posing a potential risk for nontarget organisms, including microorganisms (e.g., Icoz et al. 2008; Saxena et al. 1999; Saxena and Stotzky 2000; Zwahlen et al. 2003).
When assessing the ecological risks of transgenic plants, their impact on soil microbes should be considered, because soil microbial communities carry out complex processes that are of major ecological and agricultural significance (e.g., for biogeochemical cycles and soil fertility; Icoz et al. 2008; Saxena et al. 1999). Any changes, however small, in the composition of the microbial community should be considered as an early warning indicator for risk assessment. There have been a number of studies, using different parameters and techniques, on the effects of Bt corn hybrids on the soil microbial community. Most studies have indicated that Bt corn causes no or only minor changes in microbial community structure, and that these changes are often transient in duration (Blackwood and Buyer 2004; Brusetti et al. 2004; Devare et al. 2004; Flores et al. 2005; Icoz and Stotzky 2008; Lang et al. 2006; Icoz et al. 2008; Saxena and Stotzky 2001a; Zwahlen et al. 2007). For example, Saxena and Stotzky (2001a) found no significant differences in the number of colony-forming units of culturable bacteria (including actinomycetes) and fungi or in the numbers of nematodes and protozoa between rhizosphere soil of Bt and non-Bt corn or between soil amended with biomass of Bt and non-Bt corn. A 4-consecutive-year study (2003–2006) of corn cultivation reported no constant statistically significant differences in the numbers of different groups of microorganisms, the activities of enzymes, or the pH between soils planted with Bt and non-Bt corn (Icoz et al. 2008). The numbers and types of microorganisms and enzyme activities differed with the season and with the variety of corn, but these differences were not related to the presence of Cry proteins in the soil (Icoz et al. 2008). Zwahlen et al. (2007) found no major changes in the decomposition of Bt corn residue or in the composition of the soil organism community. No constant significant differences in bacterial counts were detected either in a greenhouse study comparing Bt- and non-Bt-corn (Brusetti et al. 2004). As expected, major differences were present in the bacterial community in the bulk soil compared to the rhizosphere; these differences were, however, unrelated to the specific hybrid. In the study of Brusetti et al. (2004), differences were detectable only when using molecular profiling techniques, whereas conventional culturing techniques reveal no differences.
By contrast, some studies have indicated that Bt corn affects microbial communities, the activities of some enzymes, and microbe-mediated processes and functions in soil (e.g., Dinel et al. 2003; Castaldini et al. 2005; Griffiths et al. 2005, 2006; Icoz et al. 2008; Turrini et al. 2004; Xue et al. 2005). Xue et al. (2005) found that the ratio of Gram-positive to Gram-negative bacteria was lower in soil with Bt corn than in soil with near-isogenic non-Bt corn. Turrini et al. (2004) reported that root exudates of Bt corn (event 176) significantly reduced presymbiotic hyphal growth of the arbuscular mycorrhizal fungus, Glomus mosseae, compared with root exudates of another Bt hybrid (event Bt11) and non-Bt corn. Constant significant differences were also detected between Bt corn (Bt11 and Bt176) and non-Bt corn plants in both total and metabolically active 16S rRNA fractions of culturable rhizosphere heterotrophic bacteria by denaturing gradient gel electrophoresis (DGGE) and in mycorrhizal colonization by microscopy (i.e., a significantly lower level of G. mosseae was detected in roots of Bt corn; Castaldini et al. 2005). Moreover, plant residues of transgenic corn affected soil respiration, bacterial community, and mycorrhizal establishment by indigenous endophytes (Castaldini et al. 2005).
From the abundance of data available, it is clear that the effect of transgenic plants on the soil microbial community, and consequently on the evolution of potential adverse effects, depends strongly on the particular plants, techniques, protein and conditions considered (Brusetti et al. 2004). It is therefore important to have reliable tests to evaluate these effects. Some methodological approaches, including the use of molecular biological techniques, show some promise in helping to understand the impact of GM crops on soil microbial ecology (Bruinsma et al. 2003; Sanvido et al. 2007). These molecular techniques, including polymerase chain reaction (PCR)-dependent approaches, yield fingerprint-type data, which allow the complexity of a microbial community to be described.
In this study, we carried out a greenhouse experiment with the aim of gaining knowledge for the evaluation of the environmental impact of Bt corn on soil ecosystems. We assessed the effects on the bacterial and fungal community structures in soils amended with growing plants of Bt corn (Pioneer 34B24 and Nongda 1246*1482) at different stages of the life cycle by using (1) PCR-DGGE of 16S and 18S rRNA genes, and (2) cloning and determination of the nucleotide sequence of nearly complete 16S and 18S rRNA genes amplified by PCR. In addition, the effects of plant residues (leaves plus stalks) of Bt corn (Pioneer 34B24 and Nongda 61) on the microbial community were further investigated using the same molecular parameters.
Materials and methods
Soils
Soils were collected from the top layer (0–20 cm) of a conventional sweetcorn field at the Agricultural Experiment Station of South China Agricultural University, Guangzhou, China. The soil was air-dried at room temperature, homogenized by sieving (2-mm mesh), and stored at 4°C before use. The soil at this site was a clay loam, containing 18.2 g kg−1 organic matter, 0.9 g kg−1 total N, 1.6 g kg−1 total P, and 20.1 g kg−1 total K (K2O), with a pH (H2O) of 6.16 and a cation exchange capacity (CEC) of 9.54 cmol kg−1.
Experimental design
Rhizospheric microbial community structure in soils with growing Bt corn
Two transgenic Bt corn hybrids [Pioneer 34B24 (event Mon810, expressing Cry1Ab protein) from Pioneer International USA, and Nongda 1246*1428 (Cry1A) from China Agricultural University] and their near-isolines (Pioneer 34B23, and Nongda 3138) were used in the experiment. Seeds of each hybrid were planted at 75 cm spacing into a 3.0 m × 3.4 m plot (a total of four plots) in a greenhouse on 12 October 2002, watered every 3 days and fertilized five times (on 28 October; 11, 21, 30 November; and 20 December) as top spray 20 g compound fertilizer per time per pot.
Sampling (three replicates) of the rhizospheric soils from each plot was performed after 25 (nine leaves per plant), 39 (stem elongation phase), 53 (tasseling), and 82 (flowering, anthesis) days of plant growth. Plants were gently removed and the rhizospheric soils were collected by gently shaking the roots to dislodge small clumps of soil adhering to the roots. Soil samples were stored immediately at −80°C before assay.
Effect of plant residues on soil microbial communities
Seeds of Bt-transgenic corn [Pioneer 34B24, and Nongda 61 (Cry1A) from China Agricultural University] and their non-Bt isolines (Pioneer 34B23, and Nongda 3138) were planted in 31 cm × 31 cm pots (one seed per pot) on 9 June 2002, following the method described above. In October 2002, after the corn had reached physiological maturity, plants from pots representing the four hybrids were removed. Straw (leaves plus stalks) was cut into 2–4 cm length pieces, freeze-dried, ground, and sieved through a 2-mm mesh. General characteristics of the straw are listed in Table 1.
Soil samples (250 g) were mixed thoroughly with 19.05 g straw from each corn hybrid, and saturated with distilled water. Three replicates were prepared for each treatment. The blended soils were incubated in an artificial growth chamber followed Sims and Holden’s protocol (1996), with a relative humidity of 80% at 25°C. On day 15 and day 75 after incubation, the soils (three replications per treatment per time) were ground, sieved through a 2-mm sieve, and stored at −80°C until use.
Polymerase chain reaction–denaturing gradient gel electrophoresis
Isolation of soil microbial DNA
Total soil DNA was extracted using soil DNA isolation kits according to the manufacturer’s instructions (FASTDNA SPIN Kit for Soil, BIO 101, Vista, CA; Borneman et al. 1996). The final concentration of each DNA sample was adjusted to 10 ng μL−1 for all PCR.
PCR for amplification of microbial 16S rRNA gene fragments
Partial 16S rRNA genes were amplified from the extracted genomic DNA by PCR using a PTC 100 thermal cycler (MJ Research, Watertown, MA). Each PCR mixture (a total volume of 50 μL) contained 50 ng genomic DNA, 200 μM each deoxynucleotide triphosphate, 50 pmol universal primers, 2.0 mM MgCl2, 5 μL 10 × PCR buffer, and 5 units Taq DNA polymerase. The 16S rRNA genes of the bacteria were amplified with the 40-nucleotide guanine-cytosine (GC)-clamp primer PRBA338F-GC [5′-(CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG) ACTCCTACGGGAGGCAGCAG-3′] and the PRUN518R (5′-ATTACCGCGGCTGCTGG-3′; Nakatsu et al. 2000) using a thermocycling program consisting of a 5-min initial denaturation at 94°C, 30 cycles of 92°C for 30 s, 55°C for 30 s, and 72°C for 30 s, followed by 15 min at 72°C. For amplification of fungal 18S rRNA genes, the universal primers were FR1-GC [5′-(CCCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCGCGGGCC) AI*CCATTCAATCGGTAIT-3′] and FF390 (5′-CGATAACGAACGAGACCT-3′; Vainio and Hantula 2000) with the following cycling conditions: denaturation at 95°C for 8 min, 30 cycles of 95°C for 30 s, 50°C for 45 s, and 72°C for 2 min, followed by a final extension for 10 min at 72°C. All PCR products were incubated at 4°C until processed further.
Denaturing gradient gel electrophoresis
DGGE was performed on a D-Gene apparatus (Bio-Rad, Hercules, CA). Samples containing approximately equal amounts of PCR amplicons were loaded onto 8% (w/v) polyacrylamide gels (37.5:1, acrylamide:bisacrylamide) in 0.53 Tris-acetate-EDTA (TAE) with a denaturing gradient ranging from 40% to 60% denaturant for bacterial 16S rRNA genes [100% denaturant contains 7 M urea and 40% (v/v) formamide in 0.53 TAE], or ranging from 45% to 60% for fungal PCR products. Electrophoresis was performed at a constant 80 V for 14 h (bacteria) or at 50 V for 18 h (fungi) at 65°C. Following electrophoresis, the gels were rinsed and stained for 20 min in an ethidium bromide solution (0.5 mg L−1), followed by 1 min of destaining in water repeated three times. The DGGE profile images were digitally captured and recorded (Gel DocTM XR170-8170 Molecular Imager System, Bio-Rad).
DGGE data analysis
DGGE fingerprints were converted to binary data on the basis of the migration and presence of bands using the BioNumerics 2.0 software (Applied Maths, Sint-Martens-Latem, Belgium). The microcommunity of soils inferred from DGGE profiles was classified using Two-Way Indicator Species Analysis (TWINSPAN®; Hill, 1979). Detrended correspondence analysis (DCA; Hill and Gauch 1980) was used to check the delimitation defined by TWINSPAN and to study interrelationships. All statistical analyses were implemented in TWINSPAN and the Canoco for Windows v.4 software package (ter Braak and Šmilauer 1998).
Sequence analysis of the16S and 18S rRNA genes
Recovery of bands from DGGE gels
DGGE bands were picked up using sterile tips, placed in a 1.5 mL microcentrifuge tube with 20 μL sterile distilled water, and incubated overnight at 4°C. To check the purity of the extracted products, PCR was performed with the primers noted above (i.e., PRBA338F-GC and PRUN518R for bacterial genes, and FR1-GC and FF390 for fungal genes), and DGGE was carried out using previous PCR products as molecular weight markers. Only those extracted products whose reamplicons presented a single band with the same migration distance as the marker were chosen for nested PCR. The nested universal primers with non-GC-clamp [PRBA338F(5′-ACTCCTACGGGAGGCAGCAG-3′) and PRUN518R for bacterial 16S rRNA gene, and FR1(5′-AICCATTCAATCGGTAIT-3′) and EF390 for fungi 18S rRNA gene)] were applied using the same protocol as described above.
Cloning and sequencing
The nested PCR products were purified with a PCR purification kit (Guoling Company, China) and ligated into the cloning vector pGEM-T (Promega) following the manufacturer’s instructions. Ligated DNA was transformed into competent Escherichia coli DH5α cells. Plasmid inserts were extracted by the alkaline lysis method (Sambrook et al. 1989). Three to five positive clones from each DGGE band were selected randomly for sequencing with an automated ABI 3100 sequencer using a T7 primer.
Nucleotide sequences obtained in this study were compared with sequences from the GenBank database using the BLASTn program. To reduce the size of the dataset, only those reference sequences with highest similarity were selected for analysis.
Results
Microbial community in rhizosphere soils
Bacterial community
The results provided by TWINSPAN and DCA from the DGGE dataset of bacterial communities from rhizosphere soils are shown in Fig. 1. A total of 48 samples were classified into four groups by TWINSPAN analysis. However, none of the groups included all the samples from the same sampling time or the same plant hybrid or variety. Separation between Bt and non-Bt rhizosphere soils was not obviously detected by the DCA, showing that there was no great variation between samples from Bt and non-Bt hybrids. However, a small amount of variation was seen between plant individuals (replicates), indicating the spatial heterogeneity of the microenvironment. The first two axes of the DCA ordination of DGGE data described 42.2% of the total variation in bacterial community structure.

Detrended correspondence analysis (DCA) of 16S rDNA denaturing gradient gel electrophoresis (DGGE) profiles of bacterial communities of rhizosphere soils. Groups are identified according to two-way indicator species analysis (TWINSPAN) results. The numbers 1, 2, 3, and 4 adjacent to each point represent the sampling times 25 days, 39 days, 53 days, and 82 days, respectively. Sampling with three replicates was implemented. ● Pioneer 34B24(Bt), ○ Pioneer 334B23(non-Bt), ▲ Nongda 1246*1482 (Bt), △ Nongda 3138 (non-Bt)
A total of 38 operational taxonomic units (OTUs) were detected, represented by 20 DGGE bands derived from the 16S rDNA gene. Using the program BLAST, sequences with most similarity to reference strains were found in the GenBank database (Table 2). The most represented bands (i.e., those present in all samples over the sampling time) were B2 and B20 (data not shown, see Table 2 for their phylogenic affiliations). No hybrid was detected to have unique DGGE band (i.e., present only in a single hybrid). Some bands were observed in Bt or its non-Bt isoline samples only at some stages; however, the case was not constant across all of four sampling time. At some growth stages, differences in the presence of B7, B8, B9, B11, B13, B14, and B16, B17, and B19 were observed between Pioneer 34B24 and Pioneer 34B23. In addition, B6, B7, B11, B19, and B21 were differentially present in Nongda 1246*1482 and Nongda 3138 (Table 2).
Fungal community
Using TWINSPAN analysis, 48 samples were identified to four groups (Fig. 2). Similar to the results obtained for bacterial communities from rhizosphere soils, no obvious separation between Bt and non-Bt rhizosphere soil samples was detected by DCA. The first two axes of the DCA ordination described 37.0% of the total variation in the fungal community.

DCA of 18S rDNA DGGE profiles for fungal community of rhizosphere soils. Groups are identified according to TWINSPAN results. The numbers 1, 2, 3, and 4 adjacent to each point represent the sampling times 25 days, 39 days, 53 days, and 82 days, respectively. Sampling with three replicates was implemented. ● Pioneer 34B24(Bt), ○ Pioneer 34B23(non-Bt), ▲ Nongda 1246*1482 (Bt), △ Nongda 3138 (non-Bt)
By means of sequencing the 18S rRNA genes isolated from 24 DGGE bands of rhizosphere soils, a total of 41 OTUs were determined. Band F2 was found in all samples from hybrids Pioneer 34B24 and Pioneer 34B23 across the four growth stages (data not shown, see Table 3 for their phylogenic affiliations). However, no ubiquitous band was shared by all samples of Nongda 1246*1482 and Nongda 3138 across the sampling time. At some stages, differences in the occurrence of bands F3, F5, F9, and F14 were observed between Pioneer 34B24 and Pioneer 34B23, whereas F5, F7, and F18 were found differentially present in Nongda 1246*1482 and Nongda 3138.
Microbial community in soils associated with plant residues
Bacterial community
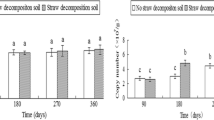
TWINSPAN analysis of bacterial communities in soils associated with plant residues distinguished two main groups differentiated by sampling time but not by plant hybrid (Fig. 3). At some sampling times, an obvious distinction between Bt and non-Bt hybrids was detected by DCA. The first two axes of the DCA ordination described 43.5% of the total variation in bacterial community structure.

DCA of 16S rDNA DGGE profiles for bacterial community of soils associated with corn straw. Groups are identified according to TWINSPAN results. The numbers 1 and 2 adjacent to each point represent sampling times 15 days and 75 days, respectively. Sampling with three replicates was implemented. ● Pioneer 34B24(Bt), ○ Pioneer 34B23(non-Bt), ▲ Nongda 61 (Bt), △ Nongda 3138 (non-Bt)
The bands that were present in all hybrids at both sampling times were B10, B14 B15, B16, B17, and B18, B19, and B20 (data not shown, see Table 2 for their phylogenic affiliations). At some sampling times, differences in the presence of some bacteria strains were revealed between Pioneer 34B24 and Pioneer 34B23, or between Nongda 61 and Nongda 3138, respectively (B1, B4, B5, B6, B7, B8, B9, B10, B14, B21, and B22).
Fungal community
The analysis of fungal communities in soils amended with plant residues by TWINSPAN and DCA led to the distinguishing of two major groups representing samples from the same sampling time (Fig. 4). The first two axes of the DCA ordination described 59.0% of the total variation in fungal community structure. Soil samples associated with Nongda 61 at 15 days were notably distinct from the others (Fig. 4).

DCA of 18S rDNA DGGE profiles for fungal communities of soils associated with corn straw. Groups are identified according to TWINSPAN results. The numbers 1 and 2 adjacent to each point represent the sampling times 15 days and 75 days, respectively. Sampling with three replicates was implemented. ● Pioneer 34B24(Bt), ○ Pioneer 34B23(non-Bt), ▲ Nongda 61 (Bt), △ Nongda 3138 (non-Bt)
Bands F3 and F8 were present in all samples from soils amended with corn straw (see Table 3). At some sampling times, differences in the presence of some bands (F2, F3, F7, F8, F11, and F12) were found between Pioneer 34B24 and Pioneer 34B23 (Table 3), whereas bands F5, F6, F7, F12, F19 were differentially present between Nongda 61 and Nongda 3138.
Discussion
Soils are home to a diverse range of life and are complex and dynamic biological systems. Therefore, it is often difficult to determine the composition of microbial communities in soils and their response to perturbations of their ecosystem (Icoz and Stotzky 2008). Recent methodological advances, especially molecular techniques, are helping researchers to understand the soil communities. In this work, we assessed the impact of growing Bt corn (Pioneer 34B24, and Nongda 1246*1482), and the straw of Bt corn (Pioneer 34B24, and Nongda 61) and the processes of decomposition on soil bacterial and fungi communities, using PCR-DGGE and sequencing of 16S and 18S rDNA. Our results proved these molecular techniques useful in revealing the diversity of microbial communities by separating different sequence variants and for identifying the affected microbial groups in the amended soils.
Effects of Bt corn on the microcommunity in rhizosphere soils
In this study, neither the bacterial nor the fungal DGGE profiles showed any obvious difference between Bt hybrid Pioneer 34B24 and non-Bt hybrid Pioneer 34B23, or between Nongda 1246*1482 (Bt) and its non-Bt isoline Nongda 3138 in the rhizosphere soils throughout the four growth stages (Figs. 1, 2). The results indicated that the root exudate of Bt corn did not constantly affect the microecosystem of the rhizosphere soil. Similar results have been reported in studies of soil microbial community structure with Cry proteins and products of other transgenic plants, e.g., Bt cotton (Gossypium hirsutum L.), Bt potato (Solanum tuberosum L.), transgenic tobacco (Nicotiana tabacum L.) expressing protease inhibitor I, transgenic alfalfa (Medicago sativa L.) expressing α-amylase or lignin peroxidase (Donegan et al. 1995, 1996, 1997, 1999). Conversely, constant significant differences have been detected in other studies of soil microbial community structure between non-transgenic and transgenic crops, such as Bt corn (Castaldini et al. 2005), Bt cotton (Gupta et al. 2002), Bt potato, etc. These conflicting results are probably due to differences in the type of Cry protein, plant variety, experimental methods used, soil type, and environmental factors, etc. (Icoz et al. 2008).
Plants can alter the composition and diversity of soil microbial communities in a selective manner (Nehl et al. 1997). The type of microbial community that results from plant-selective pressure differs with plant species, indicating that plant type and root exudates influence the microorganisms that colonize their rhizosphere (Icoz et al. 2008; Smalla et al. 2001). Indeed Bt toxin can be utilized as a carbon and nitrogen resource by soil microbes, even though its binding on clays reduced the degration. Laboratory studies by Stotzky and coworkers have indicated that the intact Cry1Ab protein binds rapidly to soil particles, maintains its insecticidal property, and persists in various soils for at least 234 days, the longest time evaluated (Crecchio and Stotzky 1998, 2001; Tapp and Stotzky 1995, 1998). Some studies reported that soil bacterial communities were more strongly influenced by plant species and different hybrids than by other environmental factors like soil type and agricultural practices (Gomes et al. 2001; Heuer et al. 2002). However, Koskella and Stotzky (2002) confirmed that several Cry toxins from B. thuringiensis have no microbicidal or microbiostatic activities against selected bacteria. Donegan et al. (1995) found that the Cry1Ab and Cry1Ac proteins, both purified and expressed in transgenic plants, had no direct effect on soil microorganisms and that the effects observed, which were related to the plant varieties used, may have been caused by unexpected changes in plant characteristics that resulted from genetic manipulation or tissue culturing of the engineered plants. Molecular data showed that both different corn lines and actively growing Bt corn plants did not reduce rhizospheric bacterial species richness, as assessed by DGGE fragments (Castaldini et al. 2005). In the present study, DCA of both bacterial and fungal communities from rhizosphere soils did not reveal any large variation between Bt lines Pioneer 34B23 with the truncated Cry1Ab gene (MON810), and Nongda 1246*1482 with the inserted Cry1A gene (Figs. 1, 2) throughout the different growth stages, indicating that the type of Cry protein, and the genetic manipulation of the engineered plants had no significant effect on soil microbial community structure.
TWINSPAN analysis inferred that plant growth stage may have a more selective effect on the microbial community than other factors, i.e., the presence of Cry protein, the type of Cry protein, and varieties used in this study (Figs. 1, 2). This result was in agreement with previous studies showing that the effects of transgenic plants on microbial communities in soil are dependent on the age of the plants. Baumgarte and Tebbe (2005) reported that, despite the detection of Cry1Ab protein in the rhizosphere of MON810 corn, the bacterial community structure was less affected by the Cry1Ab protein than by other environmental factors, e.g., the age of the plants or field heterogeneities, which was detected by single-stranded conformational polymorphism (SSCP) gel electrophoresis of 16S rRNA genes. Microbial plate counting and DGGE confirmed that differences between Bt and non-Bt corn were affected more by season (sampling time) and plant varieties, and that these differences were transient (Icoz et al. 2008). Brusetti et al. (2004) compared the rhizosphere bacterial community associated with Bt corn expressing the Cry1Ab protein and near-isogenic non-Bt corn using PCR-based automated ribosomal intergenic spacer analysis (ARISA), and found that the community structure differed with the age of the plants, suggesting that root exudates could select different bacterial communities. The relative low variability observed by ARISA in the rhizosphere samples of each plant type at 30 days could be due to the active root exudation of young plants, which could have selected a bacterial community with only a few dominant strains (Brusetti et al. 2004).
Soil type has previously been shown to be the major determinant of soil microbial community structure in agricultural systems (Girvan et al. 2003) or growth experiments (Buyer et al. 1999, 2002). It probably affects both the initial community composition and the conditions for growth during an experiment. Clay has been reported to increase the retention of Cry protein in soil, allowing for an extended period of exposure to microbes in the rhizosphere of higher clay soil (Saxena et al. 2002b; Blackwood and Buyer 2004). In this study, only one soil type, a clay roam, was implemented in all treatments. Further study comparing different soil types (e.g., clay loam, sandy clay loam, and loamy sand) will be necessary to account for whether the few significant effects from Bt corns were due to the high clay content.
Effects of plant residues of Bt corn on the soil microcommunity
Plant residues (e.g., leaves, stalks, and roots) in the corn soil represent the major reservoir of the transgenic product after harvesting, and are the primary source of carbon in soil. Therefore, any change to the quality of crop residues could modify the dynamics of the composition and activity of organisms in soil (Icoz et al. 2008). Several authors have proposed that if some microbial communities have the potential to degrade Cry1Ab, this extra protein in the environment may cause their proliferation, leading to a faster decomposition of Bt versus non-Bt maize (Blackwood and Buyer 2004; Baumgarte and Tebbe 2005; Zwahlen et al. 2007). However, biodegradation of the biomass of Bt canola, cotton, potato, rice, and tobacco was found to be significantly lower than that of the biomass of near-isogenic non-Bt plants, but the lignin content of these plant species, which was considerably lower than that of corn, was not significantly different between Bt and non-Bt biomass (Stotzky 2004).
Our previous study on the degradation of Bt protein in soils (clay loam) amended with plant straw (leaves plus stalks) of four Bt corn hybrids (Pioneer 34B24, NK58-D1, R×601 RR/ YG and Nongda 61) demonstrated a rapid decline of Bt protein in soils within the initial 14 days, and thus only a low concentration of Bt protein was detected in the soil during subsequent days (21, 30, 43, 60 and 134 days; Wang and Feng 2005). Thus, we selected 15 days and 75 days as sampling times in this study. TWINSPAN analysis of the DGGE profiles from residue-amended soil showed two major groups according to sampling time and not to the plant hybrids or varieties (Figs. 3, 4), which probably represented the result of the dynamics of degradation of plant residues (Heuer et al. 2002; Dunfield and Germida 2003; Castaldini et al. 2005).
Obvious distinction of Bt and the corresponding non-Bt hybrid detected by DCA were revealed at some sampling times (Figs. 3, 4); however, the effect was transient. Castaldini et al. (2005) found no significant differences in total soil bacterial communities affected by plant residues between Bt (Bt11, Bt176) and non-Bt lines, as inferred from the UPGMA analysis of DGGE pattern of the 16S rRNA gene. Naef et al. (2006) revealed no direct effect of the Cry1Ab protein in corn residues on the pathogen Fusarium graminearum, and on the biocontrol agent Trichoderma atroviride, and showed that some Bt and their near-isogenic non-Bt corn counterparts differed more in the chemical composition of the corn tissue as a result of different environmental conditions, such as drought-stress, which can affect the saprophytic growth of fungi on crop residues, than from the Cry protein content alone.
The effects of crop residue composition and the importance of several indices of residue quality—such as the C to N ratio, lignin content, lignin to N ratio, initial N content, initial soluble C concentrations of the residue—on decomposition and N mineralization have been extensively examined under greenhouse or field conditions (e.g., Fernandes et al. 1997; Saxena and Stotzky 2001b; Motavalli et al. 2004; Blackwood and Buyer 2004). Tarkalson et al. (2008) showed there were differences in total C, total N, biomass fractions, and C:N ratios between initial Bt (Pioneer 34N44 Bt and NC+ 4990 Bt) and non-Bt (Pioneer 34N43 and NC+ 4880) corn residues, and between companies (NC+and Pioneer); however, these differences did not result in differences in decomposition rates over time. The order of C:N ratio from the initial plant parts was leaves < stalks < cobs. Nevertheless, plant parts differed in decomposition rate where leaves > stalks > cobs. In our study, significant higher total C, total N, and C:N ratios were found in Nongda 61 (Bt) (Table 1), but this difference did not have any durable effect on soil microbial community structure (Figs. 3, 4).
In summary, PCR-DGGE and sequences of 16S rRNA and 18S rRNA genes in this study showed that Bt corn plants and their straw had no apparent lasting effect on soil bacterial and fungal communities. The age of the actively growing plants, or the time required for plant straw decomposition may have more effect on the microbial community than other factors, e.g., the presence of Cry protein, plant hybrid and variety. Although PCR-DGGE is a convenient and useful tool to track microbial communities, this method cannot provide precise quantitative information. Further studies using other experimental methods, and also more soil types, are necessary to evaluate the long-term impact of Bt corn on soil microbial communities.
References
Baumgarte S, Tebbe CC (2005) Field studies on the environmental fate of the Cry1Ab Bt-toxin produced by transgenic maize (MON810) and its effect on bacterial communities in the maize rhizosphere. Mol Ecol 14:2539–2551
Benedict JH, Ring DR (2004) Transgenic crops expressing Bt proteins: current status, challenges and outlook. In: Koul O, Dhaliwal GS (eds) Transgenic crop protection: concepts and strategies. Science, Enfeld, pp 15–83
Blackwood CB, Buyer JS (2004) Soil microbial communities associated with Bt andnon-Bt corn in three soils. J Environ Qual 33:832–836
Borneman J, Skroch PW, O'Sullivan KM, Plus JA, Rumjanek NG, Jansen JL, Nienhuis J, Triplett EW (1996) Molecular microbial diversity of an agricultural soil in Wisconsin. Appl Environ Microbiol 62:1935–1943
Bruinsma M, Kowalchuk GA, vanVeen JA (2003) Effects of genetically modified plants on microbial communities and processes in soil. Biol Fertil Soil 37:329–337
Brusetti L, Francia P, Bertolini C, Pagliuca A, Borin S, Sorlini S, Abruzzese A, Sacchi G, Viti C, Giovannetti L, Giuntini E, Bazzicalupa M, Daffonchio D (2004) Bacterial communities associated with the rhizosphere of transgenic Bt 176 maize (Zea mays) and its non-transgenic counterpart. Plant Soil 266:11–21
Buyer JS, Roberts DP, Russek-Cohen E (1999) Microbial community structure and function in the spermosphere as affected by soil and seed type. Can J Microbiol 45:138–144
Buyer JS, Roberts DP, Russek-Cohen E (2002) Soil and plant effects on microbial community structure. Can J Microbiol 48:955–964
Castaldini M, Turrini A, Sbrana C, Benedetti A, Marchionni M, Mocali S, Fabiani A, Landi S, Santomassimo F, Pietrangeli B, Nuti MP, Miclaus N, Giovannetti M (2005) Impact of Bt corn on rhizospheric and soil eubacterial communities and on beneficial mycorrhizal symbiosis in experimental microcosms. Appl Environ Microbiol 71:6719–6729
Crecchio C, Stotzky G (1998) Insecticidal activity and biodegradation of the toxin from Bacillus thuringiensis subsp. kurstaki bound to humic acids from soil. Soil Biol Biochem 30:463–470
Crecchio C, Stotzky G (2001) Biodegradation and insecticidal activity of the toxin from Bacillus thuringiensis subsp. kurstaki bound on complexes of montmorillonite-humic acids-Al hydroxypolymers. Soil Biol Biochem 33:573–581
Devare MH, Jones CM, Thies JE (2004) Effect of Cry3Bb transgenic corn and tefluthrin on the soil microbial community. J Environ Qual 33:837–843
Dinel H, Schnitzer M, Saharinen M, Meloche F, Pare T, Dumontet S, Lemee L, Ambles A (2003) Extractable soil lipids and microbial activity as affected by Bt and non-Bt maize grown on a silty clay loam soil. J Environ Science Health 38:211–219
Donegan KK, Palm CJ, Fieland VJ, Porteous LA, Ganio LM, Schaller DL, Bucao LQ, Seidler RJ (1995) Changes in levels, species and DNA fingerprints of soil microorganisms associated with cotton expressing the Bacillus thuringiensis var. kurstaki endotoxin. Appl Soil Ecol 2:111–124
Donegan KK, Shaller DL, Stone JK, Ganio LM, Reed G, Hamm PB, Seidler RJ (1996) Microbial populations, fungal species diversity, and plant pathogen levels in field plots of potato plants expressing the Bacillus thuringiensis var. tenebrionis endotoxin. Transgenic Res 5:25–35
Donegan KK, Seidler RJ, Fieland VJ, Schaller DL, Palm CJ, Ganio LM, Cardwell DM, Steinberger Y (1997) Decomposition of genetically engineered tobacco under field conditions: persistence of the proteinase inhibitor I product and effects on soil microbial respiration and protozoa, nematode, and microarthropod populations. J Appl Ecol 34:767–777
Donegan KK, Seidler RJ, Doyle JD, Porteous LA, Digiovanni G, Widmer F, Watrud LS (1999) A field study with genetically engineered alfalfa inoculated with recombinant Sinorhizobium meliloti: effects on the soil ecosystem. J Appl Ecol 36:920–936
Dunfield KE, Germida JJ (2003) Seasonal changes in the rhizosphere microbial communities associated with field-grown genetically modified canola (Brassica napus). Appl Environ Microbiol 69:7310–7318
Fernandes ECM, Motavalli PP, Castilla C, Mukurumbira L (1997) Management control of soil organic matter dynamics in tropical land use systems. Geoderma 79:49–67
Flores S, Saxena D, Stotzky G (2005) Transgenic Bt plants decompose less in soil than non-Bt plants. Soil Biol Biochem 37:1073–1082
Girvan MS, Bullimore J, Pretty JN, Osborn AM, Ball AS (2003) Soil type is the primary determinant of the composition of the total and active bacterial communities in arable soils. Appl Environ Microbiol 69:1800–1809
Gomes NCM, Heuer H, Schonfeld J, Costa R, Mendonca-Hagler L, Smalla K (2001) Bacterial diversity of the rhizosphere of maize (Zea mays) grown in tropical soil studied by temperature gradient gel electrophoresis. Plant Soil 232:167–180
Griffiths BS, Caul S, Thompson J, Birch ANE, Scrimgeour C, Andersen MN, Cortet J, Messan A, Sausse C, Lacroix B, Krogh PH (2005) A comparison of soil microbial community structure, protozoa and nematodes in field plots of conventional and genetically modified maize expressing the Bacillus thuringiensis Cry1Ab toxin. Plant Soil 275:135–146
Griffiths BS, Caul S, Thompson J, Birch ANE, Scrimgeour C, Cortet J, Foggo A, Hackett CA, Krogh PH (2006) Soil microbial and faunal community responses to Bt maize and insecticide in two soils. J Environ Qual 35:734–741
Gupta VVSR, Roberts GN, Neate SM, McClure SG, Crisp P, Watson SK (2002) Impact of Bt-cotton on biological processes in Australian soils. In: Akhurst RJ, Beard CE, Hughes PA (eds) Proceedings of the Fourth Pacific Rim Conference on the Biotechnology of Bacillus thuringiensis and Its Environmental Impacts. CSIRO, Australia, pp 191–194
Heuer H, Kroppenstedt RM, Lottmann J, Berg G, Smalla K (2002) Effects of T4 lysozyme release from transgenic potato roots on bacterial rhizosphere communities are negligible relative to natural factors. Appl Environ Microbiol 68:1325–1335
Hill MO (1979) TWINSPAN—a FORTRAN program for arranging multivariate data in an orderd two-way table classification of the individuals and attributes. Cornell University, Ithaca, NY
Hill MO, Gauch HG Jr (1980) Detrended correspondence analysis, an improved ordination technique. Vegetatio 42:47–58
Icoz I, Stotzky G (2007) Cry3Bb1 protein from Bacillus thuringiensis in root exudates and biomass of transgenic corn does not persist in soil. Transgenic Res 17:609–620, http://www.springerlink.com/content/pm447n1340n136t3
Icoz I, Stotzky G (2008) Fate and effects of insect-resistant Bt crops in soil ecosystems. Soil Biol Biochem 40:559–586
Icoz I, Saxena D, Andow DA, Zwahlen C, Stotzky G (2008) Microbial populations and enzyme activities in soil in situ under transgenic corn expressing Cry proteins from Bacillus thuringiensis. J Environ Qual 37:647–662
James C (2005) Executive summary of global status of commercialized. Biotech/GMCrops. SAAA Briefs No.34, Ithaca, NY. http://www.isaaa.org
Koskella J, Stotzky G (2002) Larvicidal toxins from Bacillus thuringiensis subspp. kurstaki, morrisoni (strain tenebrionis), and israelensis have no microbicidal or microbiostatic activity against selected bacteria, fungi, and algae in vitro. Can J Microbiol 48:262–267
Lang A, Arndt M, Beck R, Bauchhenss J, Pommer G (2006) Monitoring of the environmental effects of the Bt gene. Bavarian State Research Center for Agriculture No.2006/10, Freising-Weihenstephan. http://www.LfL.bayern.de
Lehman RM, Osborne SL, Rosentrater KA (2008) No differences in decomposition rates observed between Bacillus thuringiensis and non- Bacillus thuringiensis corn residue incubated in the field. Agron J 100:163–168
Motavalli PP, Kremer RJ, Fang M et al (2004) Impact of genetically modified crops and their management on soil microbially mediated plant nutrient transformations. J Environ Qual 33:816–824
Naef A, Zesiger T, Defago G (2006) Impact of transgenic Bt maize residues on the mycotoxigenic plant pathogen Fusarium graminearum and the biocontrol agent Trichoderma atroviride. J Environ Qual 35:1001–1009
Nakatsu CH, Torsvik V, Øvreås L (2000) Soil community analysis using DGGE of 16S rDNA polymerase chain reaction products. Soil Sci Soc Am J 64:1382–1388
Nehl DB, Allen SJ, Brown JF (1997) Deleterious rhizosphere bacteria: an integrating perspective. Appl Soil Ecol 5:1–20
Sambrook JE, Fritsch F, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Sanvido O, Romeis J, Bigler F (2007) Ecological impacts of genetically modified crops: ten years of field research and commercial cultivation. Adv Biochem Eng Biotechnol 107:235–278
Saxena D, Stotzky G (2000) Insecticidal toxinfrom Bacillus thuringiensis is released from roots of transgenic Bt corn in vitro and in situ. FEMS Microbiol Ecol 33:35–39
Saxena D, Stotzky G (2001a) Bacillus thuringiensis (Bt) toxin released from root exudates and biomass of Bt corn has no apparent effect on earthworms, nematodes, protozoa, bacteria, and fungi in soil. Soil Biol Biochem 33:1225–1230
Saxena D, Stotzky G (2001b) Bt corn has a higher lignin content than non-Bt corn. Am J Bot 88:1704–1706
Saxena D, Flores S, Stotzky G (1999) Transgenic plants: insecticidal toxin in root exudates from Bt corn. Nature 402:480
Saxena D, Flores S, Stotzky G (2002a) Bt toxin is released in root exudates from 12 transgenic corn hybrids representing three transformation events. Soil Biol Biochem 34:133–137
Saxena D, Flores S, Stotzky G (2002b) Vertical movement in soil of insecticidal Cry1Ab protein from Bacillus thuringiensisi. Soil Biol Biochem 34:111–120
Saxena D, Stewart CN, Altosaar I, Shu Q, Stotzky G (2004) Larvicidal Cry proteins from Bacillus thuringiensis are released in root exudates of transgenic B.thuringiensis corn, potato, and rice but not of B.thuringiensis canola, cotton, and tobacco. Plant Physiol Biochem 42:383–387
Sims SR, Holden LR (1996) Insect bioassay or determining soil degradation of Bacillus thuringiensis ssp. kurstaki Cry1Ab protein in corn tissue. Environ Entomol 25:659–664
Smalla K, Wieland G, Buchner A, Zock A, Parzy J, Kaiser S, Roskot N, Heuer H, Berg G (2001) Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Appl Environ Microbiol 67:4742–4751
Stotzky G (2004) Persistence and biological activity in soil of the insecticidal proteins from Bacillus thuringiensis, especially from transgenic plants. Plant Soil 266:77–89
Tapp H, Stotzky G (1995) Insecticidal activity of the toxin from Bacillus thuringiensis subspecies kurstaki and tenebrionis adsorbed and bound on pure and soil clays. Appl Environ Microbiol 61:1786–1790
Tapp H, Stotzky G (1998) Persistence of the insecticidal toxin from Bacillus thuringiensis subsp. kurstaki in soil. Soil Biol Biochem 30:471–476
Tarkalson DD, Kachman SD, Knops JMN, Thies JE, Wortmann CS (2008) Decomposition of Bt and non-Bt corn hybrid residues in the field. Nutr Cycl Agroecosyst 80:211–222
Ter Braak CFJ, Šmilauer P (1998) CANOCO Reference manual and user’s guide to Canoco for windows. Software for Canonical Community Ordination (Version 4). Centre for Biometry, Wageningen
Turrini A, Sbrana C, Nuti MP, Pietrangeli B, Giovannetti M (2004) Development of amodel system toassess the impact of genetically modifed corn and aubergine plants on arbuscular mycorrhizal fungi. Plant Soil 266:69–75
Vainio EV, Hantula J (2000) Direct analysis of wood-inhabiting fungi using denaturing gradient gel electrophoresis of amplied ribosomal DNA. Mycol Res 104:927–936
Wang JW, Feng YJ (2005) Degradation of Bt protein in soil released from Bt corn stalks and comparison of its simulation models. Chin J Ecol 24:1063–1067
Wu K (2007) Monitoring and management strategy for Helicoverpa armigera resistance to Bt cotton in China. J Invertebrate Pathol 95:220–223
Xue K, Luo HF, Qi HY, Zhang HX (2005) Changes in soil microbial community structure associated with two types of genetically engineered plants analyzing by PLFA. J Environ Sci (China) 17:130–134
Zscheischler J, Estler MC, Staudacher W et al. (1984) Handbuch Mais.Anbau, Verwertung. Fütterung-Verlagsunion Agrar, Frankfurt amMain
Zwahlen C, Hilbeck A, Gugerli P, Nentwig W (2003) Degradation of the Cry1Ab protein within transgenic Bacillus thuringiensis corn tissue in the field. Mol Ecol 12:765–775
Zwahlen C, Hilbeck A, Nentwig W (2007) Field decomposition of transgenic Bt maize residue and the impact on non-target soil invertebrates. Plant Soil 300:245–257
Acknowledgments
We would like to thank Professor Cindy Nakatsu (Purdue University, West Lafayette, IN) and Professor Jingrui Dai (Chinese Agricultural University, China) for providing corn seeds for this study. We are grateful to Dr. Tao Xu and Dr. Zhong Qin for helpful advice on statistics, and for comments on this paper. We also appreciate the helpful comments on the manuscript of two anonymous referees. This study was supported by grants from National 973 project of China (2006CB100200), the National Natural Science Foundation of China (30470335, 30770402) and the Department of Science and Technology of Guangdong, China (2006B50104002, 2007A020300009-1), the Natural Science Foundation for Doctoral Program of Guangdong province (8451064201001009), and the President Fund of South China Agricultural University (2007K037, 2008K021).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Juha Mikola.
Fengxiao Tan and Jianwu Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Tan, F., Wang, J., Feng, Y. et al. Bt corn plants and their straw have no apparent impact on soil microbial communities. Plant Soil 329, 349–364 (2010). https://doi.org/10.1007/s11104-009-0163-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-009-0163-2