
Abstract
Three different bacterial species are regularly described from tsetse flies. However, no broad screens have been performed to investigate the existence of other bacteria in this medically and agriculturally important vector insect. Utilising both culture dependent and independent methods we show that Kenyan populations of Glossina fuscipes fuscipes harbour a surprising diversity of bacteria. Bacteria were isolated from 72% of flies with 23 different bacterial species identified. The Firmicutes phylum dominated with 16 species of which seven belong to the genus Bacillus. The tsetse fly primary symbiont, Wigglesworthia glossinidia, was identified by the culture independent pathway. However, neither the secondary symbiont Sodalis nor Wolbachia was detected with either of the methods used. Two other bacterial species were identified with the DNA based method, Bacillus subtilis and Serratia marcescens. Further studies are needed to determine how tsetse flies, which only ever feed on vertebrate blood, pick up bacteria and to investigate the possible impact of these bacteria on Glossina longevity and vector competence.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Studies on the microbiota in insect guts have revealed that bacteria may affect their hosts in many different ways, from making a nutritional contribution to the insect to protecting it from pathogenic bacteria and the production of semiochemicals (Dillon and Dillon 2004). Several insects harbour symbionts (Baumann and Moran 1997). For instance, tsetse flies harbour two different symbiotic bacteria. First, the primary symbiont Wigglesworthia glossinidia which is an obligate mutualist and is postulated to help the fly with vitamin production (Akman et al. 2002). Second, the secondary symbiont Sodalis glossinidius which has been found to increase longevity and hence is described as beneficial to the fly (Dale and Welburn 2001). S. glossinidius has also been suggested to affect how efficient the flies are in transmitting the protozoan parasites of the genus Trypanosoma that cause sleeping sickness in humans and nagana in cattle (Geiger et al. 2007; Welburn and Maudlin 1999). In addition, tsetse flies may also harbour bacteria of the genus Wolbachia which are often described as pathogens in other insects although their function in tsetse flies remains to be determined. While W. glossinidia, as an obligate mutualist, is present in all tsetse flies the prevalence of Sodalis and Wolbachia varies considerably between fly populations. S. glossinidius has been detected in colony flies at high prevalence (Shaw and Moloo 1991), however, few studies have been performed on the prevalence of this species in field-caught flies. It has been shown that S. glossinidius is present in field populations of Glossina palpalis palpalis (9.3%), Glossina nigrofusca (85%) and Glossina pallicera (31%) in Liberia, however very few flies were examined for the last two species (Maudlin et al. 1990). In contrast, S. glossinidius was found in all Glossina p. gambiensis flies examined in Burkina Faso, however, in that study pupae from field caught females were investigated instead of screening the field caught flies directly (Geiger et al. 2007). S. glossinidius can be grown on nutrient agar plates under specialised conditions (Matthew et al. 2005), however neither W. glossinidia nor Wolbachia from tsetse flies have been grown in vitro. In fact, it has been suggested that the majority (80–99%) of all bacteria cannot grow under standard culturing conditions (Ward et al. 1990; Amann et al. 1995). DNA based techniques have revolutionised studies on bacterial diversity in nature, including studies on insect microbiota (Weisburg et al. 1991; Amann et al. 1995). The 16S ribosomal RNA gene is most frequently utilised to identify bacterial species in these studies. One technique utilised to screen environmental samples for bacteria is denaturing gradient gel electrophoresis (DGGE, Muyzer and Smalla 1998) which has proved to be very useful, especially since it allows for identification of the bacterial species (Dorigo et al. 2005; Nocker et al. 2007). Previous studies on insect microbiota have highlighted the need to combine culture dependent and culture independent methods since DNA based techniques also have limits (Forney et al. 2004; Wintzingerode et al. 1997; Baker et al. 2003). For example, several or most of the bacterial species isolated on culture plates in insect microbiota screens were not identified with the culture independent methods used, which in theory should detect all bacteria present (Lindh et al. 2005; Zahner et al. 2008).
Tsetse flies (Diptera: Glossinidae) are vectors of several species of African trypanosomes pathogenic to both man and his domesticated animals. The diseases they cause continue to have a devastating impact at the level of individual lives and the development of the entire African continent (Waage 1979). Glossina fuscipes fuscipes, which is investigated in this study, is implicated as the vector of about 90% of all current sleeping sickness cases in Africa. Tsetse flies are obligate haematophages, feeding on nothing but vertebrate blood throughout their entire lives (Walshe et al. 2009). As such the fly midgut might be expected to have a very limited or biased microbiota. This merits investigation, not only because this is a very unusual niche, but also because in mosquitoes the midgut microbiota have been shown to profoundly influence susceptibility to malaria parasites (Plasmodium spp., Dong et al. 2009) and the same may be true for tsetse, where many of the most important trypanosome species first become established in the fly midgut. Recently, Geiger et al. (2009) identified bacteria other than the two previously described symbionts and Wolbachia in tsetse flies utilising a culture dependent method. The study reported here is the first screen for bacteria in tsetse-flies using both a culture dependent and a culture independent pathway.
Materials and methods
Culture dependent pathway
Flies and bacterial isolates
Glossina fuscipes fuscipes were caught in biconical traps on Chamaunga Island in Lake Victoria, western Kenya (0°25′S, 34°13′E) during 4 separate days in October and November, 2008. The flies, 39 males and 25 females, were killed at −20°C, washed in a series of three baths containing: sterile water, 70% ethanol (approx. 3 min) and sterile water again. The flies were then mashed in 200 μl of sterile saline, vortexed briefly, after which 50 μl of the solution were spread on LA-plates (Luria Broth Base, Invitrogen). Two types of controls were performed each time bacterial cultivation was performed. First, a sterility control, in which materials were treated as for the other samples, but in the absence of a fly. Second a surface bacteria control where a fly was placed in 200 μl of sterile saline and vortexed briefly (without first mashing the fly) after which 50 μl of the saline was spread on LA-plates. All colonies with different colony morphologies were re-streaked on LA-plates after approx. 24 and 48 h incubation at room temperature and re-incubated under the same conditions. The bacterial isolates were stored as glycerol stocks at −20°C for approximately 2 months before transport to the laboratory in Liverpool, UK. Once there, isolates were re-streaked and new glycerol stocks were made which were then stored at −70°C. Brief morphological descriptions of colony size, shape and colour and bacteria cell size, shape and motility were noted for each isolate. These descriptions were later used together with the DGGE data to group bacterial isolates of the same species.
PCR-denaturing gradient gel electrophoresis
A region of the 16S rRNA gene of the bacterial isolates was amplified with the following PCR primers 357f gc (5′-CGC CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG GCC TAC GGG AGG CAG CAG-3′) and 518r (5′-ATT ACC GCG GCT GCT GG-3′). A GC-clamp was attached to one of the primers (357f gc, in italics) to prevent complete strand dissociation during electrophoresis (Sheffield et al. 1989). To prepare template for PCR a single colony from a LA-plate was placed with the aid of a sterile tooth pick into a 1.5 ml Eppendorf tube with 50 μl of sterile water. The tube was placed on a heating block (95°C) for 10 min and then centrifuged (14,000 rpm) for 5 min. One μl of the supernatant was added as template to a 0.25 µl PCR reaction with 10 pmol of each primer using Ready-To-Go PCR beads, (Amersham Pharmacia Biotech). The PCR program utilised, which was optimised in a similar screen of Anopheles gambiae midgut bacteria (Lindh et al. 2005), was as follows: 94°C for 3 min followed by 30 cycles of 94°C for 30 s, 58–48°C for 30 s (temperature decreased by 1°C every cycle for 10 cycles and then held at 48°C for 20 cycles), 72°C for 1 min followed by a final extension step at 72°C for 5 min. PCR fragments were analysed by electrophoresis on agarose gels (1%) before DGGE was performed on the samples. During DGGE (performed with equipment from BioRad), PCR fragments were separated with the aid of a polyacrylamide and denaturing gradient gel from 6% acrylamide/30% denaturing to 12% acrylamide/60% denaturing where 100% denaturing gel contains 7 M Urea and 40% (v/v) formamide. The gels were run in 1× TAE buffer at 200 V for 5 h at a constant temperature of 60°C. The gels were stained for 20 min with ethidium-bromide (1 mg/ml, 1× TAE), rinsed for 20 min (1× TAE) and then visualized on a UV trans-illumination table.
Sequencing and sequence analysis
Based on DGGE and morphology results (data not shown), the isolates were grouped and a few isolates from each group were selected for sequencing. The 16S rRNA genes from the selected isolates were amplified by PCR using the forward primer 8f (5′-AGA GTT TGA TNN TGG CTC AG-3′) and the reverse primer 1401r (5′-CGG TGT GTA CAA GAC CC-3′). Template preparation, PCR conditions and the PCR program were as above. PCR products were purified using QIAquick PCR Purification Kit (Qiagen) and sequenced at Macrogen, Korea on an ABI 3700 automatic DNA sequencer with primers 8f and 800r (5′-TAC CAG GGT ATC TAA TCC-3′). Preliminary sequence analyses were performed utilizing the Ribosomal Database Project II (RDP II, http://rdp.cme.msu.edu, Cole et al. 2005) and Blastn (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). Further sequence analysis was performed with selected sequences from closely related species from RDPII using MEGA (Tamura et al. 2007).
Culture independent pathway
Flies and DNA extraction
G. f. fuscipes were caught in biconical traps on Chamaunga Island, in August and September, 2007. The flies were killed at −20°C, stored in acetone and transported to Liverpool, UK. The legs and wings were removed before the flies were mashed in 180 μl of PBS. DNA extractions were then performed according to the manufacturer’s instructions for animal tissue DNA extraction with a DNeasy Blood and Tissue kit (Qiagen). The DNA was eluted in 200 μl of buffer AE. In total, 30 males and 45 females were processed. No fly was stored in acetone for more than five months before DNA extraction.
PCR–DGGE, sequencing and analysis
The eluted DNA (1–2 μl) was used as a template in PCR reactions with the same primers and conditions as for the bacterial isolates above. Genomic DNA from G. m. morsitans and G. p. palpalis colony flies were used as controls. After determining that the 357f–518r primer pair amplifies a region of the Glossina 18S rRNA gene another reverse primer was utilised: 800r (5′-TAC CAG GGT ATC TAA TCC-3′) together with the same forward primer. All fly DNA samples were screened with both primer pairs and analysed with DGGE. Agarose electrophoresis and DGGE conditions were as above. Interesting bands from the DGGE gels were excised with the aid of a sterile pipette tip. The round gel pieces were incubated at 4°C overnight to elute the DNA. The eluted DNA was then used as template in PCR reactions. The PCR program, PCR purification, sequencing and sequence analysis were performed as above with the exception that the sequencing primers utilised were the same primers as originally used to amplify the fragment for DGGE analysis but without the gc clamp.
Detection limits for S. glossinidius and B. subtilis when extracted with G. f. fuscipes genomic DNA
A B. subtilis strain isolated during this study was grown in LB media to OD600 = 0.5 which according to viable counts performed on LA plates correspond to 3.9 × 108 colony forming units/ml. Bacterial DNA was prepared using the blood and tissue culture kit (Qiagen) from 5 ml of the bacterial suspension following the protocol for Gram positive bacteria. S. glossinidius DNA from a known number of bacterial cells were kindly provided by Lee Haines, LSTM, Liverpool, UK. DNA from the two bacterial species were serially diluted in tsetse DNA from two G. f. fuscipes flies and then used in PCR–DGGE protocols with the 357f gc–518r and the 357f gc–800r primer pairs as above.
Results
Culture dependent pathway
Bacteria were found in 46 (72%) out of the 64 flies screened with the culture dependent pathway. In total 25 different species were identified from 145 isolates of which three species were also found in the surface controls (Table 1), i.e. those three bacterial species may have originated from the outer cuticle as well as the interior organs of the flies. One species, Bacillus megaterium, was identified in one of the sterility controls and data for this species have been excluded from the results. If the species identified in the surface controls are also excluded then bacteria were identified in 42% of the flies. The majority of the isolates identified (n = 16) belong to the phylum Firmicutes and the genus with most isolates was Bacillus with seven species (Table 1; Fig. 1). The other two phyla represented in this study were Proteobacteria, with eight isolates, and Actinobacteria, from which an Arthrobacter sp. was identified (Table 1). The maximum number of bacterial species isolated from one fly was five, two of these were also found in at least one of the surface controls. If the bacterial species isolated from the surface controls are excluded two flies harboured three different bacterial species, and 10 and 15 flies harboured two and one species, respectively.

Evolutionary relationships of the Bacillus species. Lineages labelled bx where x is a number refer to the different bacterial isolates from tsetse flies that were sequenced. Bacillus cereus, Bacillus thuringiensis and Bacillus anthracis are very hard to separate based on the 16S rRNA sequence (Ash et al. 1991). The evolutionary history was inferred using the Maximum Parsimony method (Eck and Dayhoff 1966). The bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analysed (Felsenstein 1985). Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches (Felsenstein 1985). The MP tree was obtained using the Close-Neighbor-Interchange algorithm (Nei and Kumar 2000, p. 128) with search level 3 (Felsenstein 1985; Nei and Kumar 2000) in which the initial trees were obtained with the random addition of sequences (10 replicates). All positions containing gaps and missing data were eliminated from the dataset. There were a total of 667 positions in the final dataset, out of which 509 were parsimony informative. Phylogenetic analyses were conducted in MEGA4 (Tamura et al. 2007)
Culture independent pathway
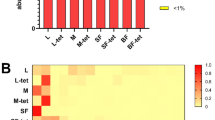
As expected, the primary symbiont W. glossinidia was found in all flies screened with the culture independent method (Fig. 2). However, neither Wolbachia nor S. glossinidius were identified in the field caught G. f. fuscipes. All three Glossina symbionts were detected in the G. m. morsitans (Genbank accession numbers: Wolbachia sp. GU361692, GU361701; S. glossinidius GU361693, GU361716: W. glossinidia GU361700, GU361714) and S. glossinidius (GU361699) and W. glossinidia (GU361694, GU361698, GU361715) were detected in the G. palpalis flies used as controls in this study (Fig. 2). Furthermore, an additional screen of 25 of the field caught G. f. fuscipes flies using Sodalis specific primers failed to detect Sodalis (data not shown). In addition to W. glossinidia (GU361695-97; GU361704-6; GU361708-9; GU361712-13), only Glossina 18S rRNA and Serratia marcescens (GU361702-3) were identified by the culture independent method with the 357gcf-518r primer pair (Fig. 2). Since the reverse primer 518r has a perfect match with the tsetse 18S rRNA gene, this primer was changed (to primer 800r). The second primer pair did not amplify the 18S gene. However, this primer pair only detected one bacterial species in addition to the tsetse symbionts and Serratia marcescens (GU361710-11). This second non symbiotic bacterial species was B. subtilis (GU361707) which, in contrast to S. marcescens, was also isolated on plates.

DGGE gel visualising bacteria in G. f. fuscipes field flies and G. m. morsitans and G. p. palpalis controls PCR with primers 357f gc and 518r. Lanes 1–8: G. f. f. females, lane 9: G. m. m. colony fly, lane 10: G. p. p. colony fly. Bands A: Glossina 18S, B: W. glossinidia, C: Wolbachia sp. D: S. marcescens E: S. glossinidius. Wigglesworthia are species specific and hence the 16S sequences vary slightly between Glossina species. S. marcescens generates two bands
The detection limit of bacterial DNA in the presence of tsetse DNA was investigated for one of the Bacillus species (B. subtilis) and for S. glossinidius. The detection limit was approximately the same for both primer pairs utilised: between 4 × 103 and 4 × 102 B. subtilis and 105 and 104 S. glossinidius bacterial cells could be detected (Fig. 3 and data not shown).

DGGE gel visualising the detection limit of B. subtilis and S. glossinidius in tsetse DNA. PCR with primers 357f gc and 800r. Lane 1: B. subtilis DNA, lanes 2–6: B. subtilis diluted in tsetse DNA, 4 × 106–4 × 102 bact./PCR react, 10× dilution between each line, lanes 7–12: S. glossinidius diluted in tsetse DNA, 106–101 bact./PCR react, 10× dilution between each line, lane 13: G. f. f. DNA used for bacterial DNA dilutions. Bands A: B. subtilis, B: S. glossinidius, C: W. glossinidia
Discussion
Culture dependent pathway
In this study Firmicutes and Bacillus were the dominant phylum and genus respectively. In contrast, during a recent study on the cultivable flora of Glossina palpalis palpalis 39 bacterial strains were isolated out of which 95% were Gram negative and 82% belonged to the Gamma-proteobacteria class (Geiger et al. 2009). Several other insect microbiota screens have also reported Gamma-proteobacteria as the dominant class (Azambuja et al. 2005; Lindh et al. 2005; Zientz et al. 2001). Seven species from the Gamma-proteobacteria class were isolated in this study, four of these belong to the family Enterobacteriaceae (Table 1; Fig. 4), where many insect symbionts are found including W. glossinidia and S. glossinidius. Many of the bacterial species identified in this study have also been detected in other insect vector species (Azambuja et al. 2005; Lindh 2007), however not in the screen of G. p. palpalis (Geiger et al. 2009). The difference in bacterial flora between G. f. fuscipes and G. p. palpalis may reflect differences in blood-feeding patterns between the two species (Mayer and James 1969). However, it may also reflect the different culture media utilised in the two studies (Geiger et al. 2009). It is possible that an even more diverse bacterial flora would have been detected on culture plates in this study if several different culture media and conditions had been utilised.

Evolutionary relationships of the gamma-proteobacteria species See legend for Fig. 1. There were a total of 653 positions in the final dataset, out of which 151 were parsimony informative. Genebank accession numbers GQ009466, GQ069739 and EU538337 belong to bacteria found on human skin (Grice et al. 2008, 2009)
The high prevalence and diversity of cultivatable bacteria in tsetse flies is surprising given that they are monophagous, only feeding on vertebrate blood throughout their life. In the main vertebrate blood is sterile. Hence the data from this study argues against the theory that polyphagous insects have a higher prevalence of intestinal bacteria and a more diverse bacterial flora (Mrazek et al. 2008; Dillon et al. 2008). It is interesting to note that bacteria were isolated from 72% of the tsetse flies in this study while a study of bacteria in Anopheles mosquito midguts performed in the same area in 2005 only detected bacteria in 15% of the mosquitoes and only in 7% with the culture dependent method utilising the same type of culture plates as in this study (Lindh et al. 2005). The reason for the higher prevalence of bacteria in tsetse flies compared to mosquitoes may depend on one of two reasons or a combination of both. First, tsetse flies might encounter and ingest more bacteria than mosquitoes. Given that adult mosquitoes feed on various sugar sources in the field in addition to blood this seems improbable. Furthermore, larval mosquitoes live in water bodies with high loads of bacteria and the adults are capable of ingesting bacteria from that water after they emerge (Lindh et al. 2008). However, mosquitoes and tsetse flies have a different mode of blood-feeding and this may account for the differences seen. Mosquitoes cannulate capillaries and so suck blood from within the enclosed vertebrate blood system. In contrast, tsetse flies have much larger mouthparts which they use to drill into the skin of the vertebrate causing a sub-dermal haematoma from which they suck blood (Lehane 2005). The sub-dermal haematoma may well be contaminated with bacteria from the skin surface. This possibility is supported by the findings of Kaaya and Darji (1989) and Poinar et al. (1979) who showed that G. m. morsitans mortality was increased after feeding on rabbit ears that had been smeared with different species of bacteria that were supposed to be harmful to the flies. Given this hypothesis it is interesting to note that two of the Enterobacteriaceae spp. isolated in this study are close relatives of bacteria found in human skin microbiota (Fig. 4, Grice et al. 2009). The second reason may be that tsetse flies have a weaker intestinal anti-bacterial immune system compared to Anopheles mosquitoes. Anopheles may need a stronger immune system since, as argued above, they imbibe a broader and more bacterially contaminated set of foods. Alternatively, tsetse flies may have an impaired anti-bacterial response to allow for symbiont survival. Yet another difference between mosquitoes and tsetse which may have a bearing here is length of adult life. Female tsetse can live for 100 days or more while adult mosquitoes rarely achieve 30 days of adult life. As both species feed every 2–3 days clearly a population of tsetse flies will have had a greater chance of contracting a bacterial load if this is associated with the feeding process.
Culture independent pathway
Only two bacterial species other than the tsetse symbiont W. glossinidia were identified from field caught G. f. fuscipes with the culture independent pathway utilising two different primer pairs. We speculated that the high copy number of W. glossinidia 16S and the tsetse 18S rRNA genes might have caused bacteria, if present in low copy number, to fail to be detected in the PCR. However, changing the reverse primer and thereby excluding amplification of the tsetse 18S rRNA gene only resulted in one more bacterial species being identified. DNA from both B. subtilis and S. glossinidius could be detected when diluted in tsetse DNA but only when present above a threshold. Hence, we cannot exclude that Sodalis and Wolbachia, in addition to other bacteria, are present in G. f. fuscipes flies in numbers lower than the detection limits for the PCR–DGGE protocol utilised here. The similar detection limits of the two primer pairs, where one set amplifies the 18S rRNA tsetse gene while the other does not, indicate that the amplification of the 18S rRNA tsetse gene did not affect the sensitivity of the PCR. However, the high copy number of the Wigglesworthia 16S rRNA gene might still explain the difference in prevalence and diversity of non-symbiotic bacteria identified with the culture dependent and culture independent pathway utilised in this study. A solution might be to use phylum or class specific primers (Muhling et al. 2008) although this will not be feasible for species within the Gamma-proteobacteria since W. glossinidia belongs to this class.
Influence of bacteria on vector competence and longevity?
Previous studies have suggested that gut bacteria may influence parasite transmission by insects (Azambuja et al. 2005; Dong et al. 2009). For instance, Pumpuni et al. (1993) found that a Gram-negative bacteria inhibited oocyst formation of the malaria parasite Plasmodium falciparum in Anopheles stephensi mosquitoes. This observation was later confirmed by Gonzalez-Ceron et al. (2003) who reported a lower infection rate in An. albimanus when Gram-negative bacteria were co-infected with P. vivax parasites. In addition, S. marcescens isolated from Rhodnius prolixus was shown to lyse the parasite Trypanosoma cruzi in vitro (Azambuja et al. 2004). Moreover, Pseudomonas fluorescens strains have also been reported to be able to lyse T. cruzi (Mercado and Colon-Whitt 1982). As a contrast the secondary symbiont in tsetse S. glossinidius has been shown to increase susceptibility of tsetse flies to trypanosomes (Welburn et al. 1993) although the relationship appears complex (Geiger et al. 2007; Geiger et al. 2005). Bacteria have also been reported to affect Glossina mortality. A number of Gram-negative and Gram-positive bacteria, among them S. marcescens, Providencia rettgeri and several Bacillus spp. induced mortality of G. m. morsitans (Kaaya and Darji 1989). Furthermore, S. marcescens have been shown to cause increased mortality in An. albimanus mosquitoes and in G. morsitans and G. pallidipes flies (Poinar et al. 1979; Gonzalez-Ceron et al. 2003). Taken together, these studies clearly suggest that gut bacteria may influence vector competence and or longevity of disease transmitting insects. Hence, it would be interesting to investigate the impact the non-symbiotic bacteria isolated in this study may have on vector competence and mortality of tsetse flies, especially since several of the bacterial species investigated in the studies above correlate with species identified in this study.
References
Akman L, Yamashita A, Watanabe H, Oshima K, Shiba T, Hattori M, Aksoy S (2002) Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat Genet 32:402–407
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial-cells without cultivation. Microbiol Rev 59:143–169
Ash C, Farrow JA, Dorsch M, Stackebrandt E, Collins MD (1991) Comparative analysis of Bacillus anthracis, Bacillus cereus, and related species on the basis of reverse transcriptase sequencing of 16S rRNA. Int J Syst Bacteriol 41:343–346
Azambuja P, Feder D, Garcia ES (2004) Isolation of Serratia marcescens in the midgut of Rhodnius prolixus: impact on the establishment of the parasite Trypanosoma cruzi in the vector. Exp Parasitol 107:89–96
Azambuja P, Garcia ES, Ratcliffe NA (2005) Gut microbiota and parasite transmission by insect vectors. Trends Parasitol 21:568–572
Baker GC, Smith JJ, Cowan DA (2003) Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55:541–555
Baumann P, Moran NA (1997) Non-cultivable microorganisms from symbiotic associations of insects and other hosts. Antonie Van Leeuwenhoek Int J Gen 72:39–48
Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM (2005) The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res 33:294–296
Dale C, Welburn SC (2001) The endosymbionts of tsetse flies: manipulating host–parasite interactions. Int J Parasitol 31:628–631
Dillon RJ, Dillon VM (2004) The gut bacteria of insects: nonpathogenic interactions. Annu Rev Entomol 49:71–92
Dillon RJ, Webster G, Weightman AJ, Dillon VM, Blanford S, Charnley AK (2008) Composition of Acridid gut bacterial communities as revealed by 16S rRNA gene analysis. J Invertebr Pathol 97:265–272
Dong Y, Manfredini F, Dimopoulos G (2009) Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog 5:e1000423
Dorigo U, Volatier L, Humbert JF (2005) Molecular approaches to the assessment of biodiversity in aquatic microbial communities. Water Res 39:2207–2218
Eck RV, Dayhoff MO (1966) Atlas of protein sequence and structure. National Biomedical Research Foundation, Silver Springs, MD
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Forney LJ, Zhou X, Brown CJ (2004) Molecular microbial ecology: land of the one-eyed king. Curr Opin Microbiol 7:210–220
Geiger A, Ravel S, Frutos R, Cuny G (2005) Sodalis glossinidius (Enterobacteriaceae) and vectorial competence of Glossina palpalis gambiensis and Glossina morsitans morsitans for Trypanosoma congolense savannah type. Curr Microbiol 51:35–40
Geiger A, Ravel S, Mateille T, Janelle J, Patrel D, Cuny G, Frutos R (2007) Vector competence of Glossina palpalis gambiensis for Trypanosoma brucei s.l. and genetic diversity of the symbiont Sodalis glossinidius. Mol Biol Evol 24:102–109
Geiger A, Fardeau ML, Grebaut P, Vatunga G, Josenando T, Herder S, Cuny G, Truc P, Ollivier B (2009) First isolation of Enterobacter, Enterococcus, and Acinetobacter spp. as inhabitants of the tsetse fly (Glossina palpalis palpalis) midgut. Infect Genet Evol 9:1364–1370
Gonzalez-Ceron L, Santillan F, Rodriguez MH, Mendez D, Hernandez-Avila JE (2003) Bacteria in midguts of field-collected Anopheles albimanus block Plasmodium vivax sporogonic development. J Med Entomol 40:371–374
Grice EA, Kong HH, Renaud G, Young AC, Bouffard GG, Blakesley RW, Wolfsberg TG, Turner ML, Segre JA (2008) A diversity profile of the human skin microbiota. Genome Res 18:1043–1050
Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA (2009) Topographical and temporal diversity of the human skin microbiome. Science 324:1190–1192
Kaaya GP, Darji N (1989) Mortality in adult tsetse, Glossina morsitans morsitans, caused by entomopathogenic bacteria. J Invertebr Pathol 54:32–38
Lehane MJ (2005) Biology of blood-sucking in insects, 2nd edn. Cambridge University Press, Cambridge
Lindh JM (2007) Identification of bacteria associated with malaria mosquitoes—their characterisation and potential use. Stockholm University, Stockholm
Lindh JM, Terenius O, Faye I (2005) 16S rRNA gene-based identification of midgut bacteria from field-caught Anopheles gambiae sensu lato and A. funestus mosquitoes reveals new species related to known insect symbionts. Appl Environ Microb 71:7217–7223
Lindh JM, Borg-Karlson AK, Faye I (2008) Transstadial and horizontal transfer of bacteria within a colony of Anopheles gambiae (Diptera: Culicidae) and oviposition response to bacteria-containing water. Acta Trop 107:242–250
Matthew CZ, Darby AC, Young SA, Hume LH, Welburn SC (2005) The rapid isolation and growth dynamics of the tsetse symbiont Sodalis glossinidius. FEMS Microbiol Lett 248:69–74
Maudlin I, Welburn SC, Mehlitz D (1990) The relationship between rickettsia-like-organisms and trypanosome infections in natural populations of tsetse in Liberia. Trop Med Parasitol 41:265–267
Mayer MS, James JD (1969) Attraction of Aedes aegypti (L.): responses to human arms, carbon dioxide, and air currents in a new type of olfactometer. Bull Entomol Res 58:629–642
Mercado TI, Colon-Whitt A (1982) Lysis of Trypanosoma cruzi by Pseudomonas fluorescens. Antimicrob Agents Chemother 22:1051–1057
Mrazek J, Strosova L, Fliegerova K, Kott T, Kopecny J (2008) Diversity of insect intestinal microflora. Folia Microbiol 53:229–233
Muhling M, Woolven-Allen J, Murrell JC, Joint I (2008) Improved group-specific PCR primers for denaturing gradient gel electrophoresis analysis of the genetic diversity of complex microbial communities. ISME J 2:379–392
Muyzer G, Smalla K (1998) Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek Int J Gen 73:127–141
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
Nocker A, Burr M, Camper AK (2007) Genotypic microbial community profiling: a critical technical review. Microb Ecol 54:276–289
Poinar GO Jr, Wassink HJ, Leegwater-van der Linden ME, van der Geest LP (1979) Serratia marcescens as a pathogen of tsetse flies. Acta Trop 36:223–227
Pumpuni CB, Beier MS, Nataro JP, Guers LD, Davis JR (1993) Plasmodium falciparum—inhibition of sporogonic development in Anopheles stephensi by Gram-negative bacteria. Exp Parasitol 77:195–199
Shaw MK, Moloo SK (1991) Comparative-study on rickettsia-like organisms in the midgut epithelial-cells of different Glossina species. Parasitology 102:193–199
Sheffield VC, Cox DR, Lerman LS, Myers RM (1989) Attachment of a 40-base-pair G+C-rich sequence (Gc-Clamp) to genomic DNA fragments by the polymerase chain-reaction results in improved detection of single-base changes. PNAS 86:232–236
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Waage JK (1979) The evolution of insect/vertebrate associations. Biol J Linn Soc 12:187–224
Walshe DP, Ooi CP, Lehane MJ, Haines LR (2009) The enemy within: interactions between tsetse, trypanosomes and symbionts. Adv Insect Physiol 37:119–175
Ward DM, Weller R, Bateson MM (1990) 16s ribosomal-RNA sequences reveal numerous uncultured microorganisms in a natural community. Nature 345:63–65
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Welburn SC, Maudlin I (1999) Tsetse–typanosome interactions: rites of passage. Parasitol Today 15:399–403
Welburn SC, Arnold K, Maudlin I, Gooday GW (1993) Rickettsia-like organisms and chitinase production in relation to transmission of trypanosomes by tsetse-flies. Parasitology 107:141–145
Wintzingerode Fv, Gobel UB, Stackebrandt E (1997) Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21:213–229
Zahner V, Lucarotti CJ, McIntosh D (2008) Application of 16S rDNA-DGGE and plate culture to characterization of bacterial communities associated with the sawfly, Acantholyda erythrocephala (Hymenoptera, Pamphiliidae). Curr Microbiol 57:564–569
Zientz E, Silva FJ, Gross R (2001) Genome interdependence in insect–bacterium symbioses. Genome Biol 2:reviews1032
Acknowledgments
We thank Lee Haines for providing Sodalis glossinidius DNA and Rod Dillon, Naomi Dyer and Stella Lehane for technical assistance, discussions and support. The study was funded by the European Commission INCO programme.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lindh, J.M., Lehane, M.J. The tsetse fly Glossina fuscipes fuscipes (Diptera: Glossina) harbours a surprising diversity of bacteria other than symbionts. Antonie van Leeuwenhoek 99, 711–720 (2011). https://doi.org/10.1007/s10482-010-9546-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-010-9546-x