
Abstract
New structurally diverse natural products are discovered when novel screening procedures are introduced or when high quality biological materials from new sources are examined in existing screens, hence it is important to foster these two aspects of novelty in drug discovery programmes. Amongst prokaryotes, actinomycetes, notably streptomycetes, remain a rich source of new natural products though it has become increasingly difficult to find such metabolites from common actinomycetes as screening ‘old friends’ leads to the costly rediscovery of known compounds. The bioprospecting strategy which is the subject of this review is based upon the premise that new secondary metabolites can be found by screening relatively small numbers of dereplicated, novel actinomycetes isolated from marine sediments. The success of the strategy is exemplified by the discovery of a range of novel bioactive compounds, notably atrop-abyssomicin C and proximicins A, B and C from Verrucosispora strains isolated from sediment samples taken from the Sea of Japan and the Raune Fjord, respectively, and the dermacozines derived from Dermacoccus strains isolated from the Challenger Deep of the Mariana Trench in the Pacific Ocean. The importance of current advances in prokaryotic systematics in work of this nature is stressed and a plea made that resources be sought to train, support and employ the next generation of actinobacterial systematists.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
New drugs, especially antibiotics, are urgently needed to counter and reverse the spread of antibiotic resistant pathogens (Talbot et al. 2006; Payne et al. 2007) and to combat life-threatening diseases such as cancer (Olano et al. 2009a). It is widely acknowledged that the most promising source of new drugs remain natural products (Bull et al. 2000; Fenical and Jensen 2006; Bull and Stach 2007), especially given the inconvenient truth that alternative strategies, such as combinatorial chemistry and fragment-based drug design, have been relatively unproductive with only one de novo combinatorial New Chemical Entity approved anywhere in the world (Newman 2008). Experience has shown that previously unknown, important natural products are found when new screening systems are introduced or when high quality biological materials from new sources are examined in existing screens (Fig. 1). It is, therefore, essential in drug discovery programmes to foster these two aspects of novelty by building upon scientific and technological developments in these areas.

Twin-tracked approach to drug discovery
The choice of bacteria for pharmacological screening programmes is a daunting one given the taxonomic diversity of cultivable prokaryotes (Bull 2004a; De Vos et al. 2009). However, this diversity is but a tiny fraction of the uncultivated prokaryotic diversity present in natural habitats (Bull et al. 2000; Bull 2004a, b; Sogin et al. 2006), a silent majority of prokaryotes which encompasses enormous genetic diversity for exploitable biotechnology (Whitman et al. 1998; Bull 2004a, b). This extensive gene pool is being sampled by the application of innovative procedures for the selective isolation of previously unknown bacteria (Fry 2004; Epstein et al. 2010), including actinomycetes (Goodfellow 2010), thereby compounding the problem outlined above.
Amongst prokaryotes, members of the order Actinomycetales, notably the genus Streptomyces, remain the richest source of natural products, including clinically useful antibiotics, antimetabolites and antitumour agents (Bérdy 2005; Newman and Cragg 2007; Olano et al. 2009a, b). Actinomycete sources account for about 45% of all microbial bioactive secondary metabolites with 7,600 of these compounds (80%) being produced by Streptomyces (Bérdy 2005). Despite this astonishing productivity, it has been predicted that only about 10% of the total number of natural products that can be synthesized by these organisms have been discovered (Watve et al. 2001). However, the key to a resurgence of interest in actinomycetes as a source of new chemical entities came from the application of genomic technologies which showed that the whole genomes of Rhodococcus sp. RHA1 (McLeod et al. 2006), Saccharopolyspora erythraea NRRL 23338 (Oliynyk et al. 2007), Salinispora tropica CNB-440 (Udwary et al. 2007), Streptomyces avermitilis MA-4680 (Ōmura et al. 2001; Ikeda et al. 2003) and ‘Streptomyces coelicolor’ A(3)2 (Bentley et al. 2002) each contained around 20 or more natural product biosynthetic gene clusters for the production of known or predicted secondary metabolites. In contrast, few, if any, such gene clusters have been detected in the genomes of other bacteria, as shown by the presence of three in Bacillus subtilis 168 (Kunst et al. 1997), four in Pseudomonas aeruginosa PA01 (Stover et al. 2000) and two in Ralstonia solanacearum GMI 1000 (Salanoubat et al. 2002).
So, once again, the focus is on actinomycetes as a source of novel, clinically significant natural products. However, it is becoming increasingly difficult to find such metabolites from common actinomycetes as screening ‘old favourites’ leads to the costly rediscovery of known compounds (Williams 2008). This problem can be met by using standard procedures for the selective isolation of novel actinomycetes from poorly studied habitats (Sembiring et al. 2000; Goodfellow et al. 2007; Okoro et al. 2009), by applying new methods for the selective isolation of rare and uncommon actinomycetes (Suzuki et al. 2001a, b; Tan et al. 2006) and by devising innovative procedures for the cultivation of specific components of previously uncultivated actinomycetes known to be present in natural habitats (Stach et al. 2003a, b; Giovanonni and Stingl 2005; Allgaier and Grossart 2006), as exemplified by the isolation of seven candidate species from lakes and ponds in temperate, subtropical and tropical climatic zones (Hahn 2009).
The traditional perception of actinobacteria as authothonous soil and freshwater organisms is being radically reviewed as it is increasingly evident that members of this phylum are among the most successful colonizers of all environments in the extremobiosphere, often occurring as the dominant population (Bull 2010). Progress has also been made in drug discovery from actinomycetes by using high-throughput screening and fermentation, metabolic profiling technologies, genome scanning, mining genomes for cryptic pathways, and combinatorial biosynthesis to generate new secondary metabolites related to existing pharmacophores (Bull and Stach 2007; Baltz 2008). Metagenomic screening of DNA from environmental samples provides an alternative way of discovering new antibiotic biosynthetic genes (Handelsman 2004; Schloss and Handelsman 2005).
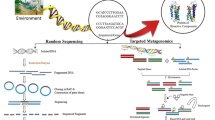
Our culture-dependent bioprospecting strategy is outlined in Fig. 2. The initial steps in this strategy are based on the use of a judicious choice of selective isolation procedures, the recognition and dereplication of target actinomycetes and on the subsequent selection of representative strains for screening. These steps are heavily dependent on developments in actinomycete systematics, based on the hypothesis that taxonomic diversity is a surrogate for chemical diversity (Ward and Goodfellow 2004; Bull and Stach 2007; Jensen 2010) and on the concept that novel species may contain unique compounds as the evolution of secondary metabolites may act as a driver for bacterial speciation (Czaran et al. 2002; Jensen 2010).

Culture-dependent bioprospecting strategy
The remaining steps in the procedure complement those outlined above, namely the expression/detection of the desired properties in dereplicated strain libraries using appropriate fermentation conditions, primary screening of fermentation broths/mycelia extracts using HPLC-diode array screening, detection of metabolite novelty using an in-house HPLC-UV-visual database and structural chemical elucidation of active principles. The final step in the process involves the full taxonomic characterization of strains giving interesting hits.
There is evidence that systematics, especially microbial systematics, is in critical decline as exemplified by a recent report from the UK House of Lords Science and Technology Committee (2008). This report paints a bleak picture of a subject which is becoming marginalised but does offer a series of recommendations to help alleviate the situation. While support for this beleaguered discipline is to be welcomed from almost any quarter it is a matter of concern that the conceptual basis of the subject is frequently misunderstood even by microbiologists. Consequently, the underlying principles of prokaryotic systematics are briefly touched upon here with particular reference to actinobacteria. More detailed and expansive consideration of the discipline can be found elsewhere (Priest and Goodfellow 2000; Brenner et al. 2005; Schleifer 2010).
Roots of prokaryotic systematics
Prokaryotic systematics is the scientific study of the kinds, diversity, and relationships within and between Archaea and Bacteria. The subject is usually divided into three separate, sequential, but interrelated subdisciplines, namely classification, nomenclature and identification. The initial step, classification, is the process of ordering organisms into taxonomic groups (taxa) on the basis of similarities and differences. The outcome is an orderly arrangement or system that is designed to show natural relationships between taxa and to serve as an information storage and retrieval system. The term classification encompasses both the process and the outcome of the exercise though outcomes are often referred to as taxonomies. Sound classification of prokaryotes is a prerequisite for stable nomenclature and reliable identification procedures.
Classifications based on large suites of genotypic and phenotypic properties are termed phenetic. This approach encompasses measurable features of prokaryotes (e.g. biochemical, chemical, morphological and physiological properties), including genetic relationships (e.g. DNA:DNA homology values). Phenetic classifications show relationships between organisms as they exist now, that is, without reference to evolutionary pathways or ancestry. In contrast, phylogenetic classifications express inferred evolutionary relatedness between organisms and thereby reflect the extent of change over time. In practice, phylogenetic classifications are usually found to be phenetically coherent. Current approaches to prokaryotic classification based on 16S rRNA gene sequences purport to be phylogenetic, but many are in fact phenetic measures of affinity with homologous nucleotide sequences as characters.
The second step, nomenclature, deals with the terms used to denote ranks in the taxonomic hierarchy (e.g. species, genera, families) and with the practice of assigning the correct, internationally recognized names to taxonomic groups according to rules laid out in successive editions of the International Code of Nomenclature of Bacteria (Lapage et al. 1975, 1992). Two reforms in the ‘Bacteriological Code’ edited by Lapage and his colleagues in 1975 have had far reaching impacts on the nomenclature of prokaryotes.
-
A definitive document and starting date for the recognition of names introduced with the publication of the Approved Lists of Bacterial Names on January 1, 1980 (Skerman et al. 1980). Names published prior to this date and omitted from the Approved Lists lost their standing in nomenclature, a development that cleared away thousands of meaningless names. Old names can be resurrected if the system for doing so is followed.
-
Names of new taxa can only be validly published in the International Journal of Systematic and Evolutionary Microbiology (IJSEM; formerly the International Journal of Systematic Bacteriology), but can be effectively published in appropriate international journals and then cited in Validation Lists published in the IJSEM.
These changes mean that the IJSEM serves as a convenient ‘one-stop-shop’ for the recognition of validly described new names of species, genera and other taxonomic ranks.
A principle of paramount importance in nomenclature and identification is the nomenclatural type concept. A taxon in the taxonomic hierarchy up to class may contain a number of elements. The elements of species are strains and those of a genus are species and so on. The nomenclatural type of a taxon is that element with which the name of a taxon is permanently associated. The type species of a genus, for instance, must be retained in the genus even if all other species are removed from it. A type, therefore, is the nominifer or name bearer, it is a reference point for the name in question.
The type of a taxon does not have any physical existence above the rank of species, it is merely a name. In contrast, at the species and subspecies level the nomenclatural type is represented by a particular strain, the type strain, which does have a physical existence, as any number of subcultures. Type strains are designated by taxonomists who describe new species. They are the permanent living embodiments of validly described species and have to be deposited in two service culture collections in different countries, so that they are readily available for study. Type strains are of the greatest importance for taxonomic work as they are reference points when attempting to identify unknown microorganisms. The knowledge that type strains may not be entirely typical of a species is outweighed by the fact that by definition they are authentic.
The correct use of names is central to all aspects of the microbial sciences as microbiologists need to know which organisms they are studying before they can pass on information about them within and outwith the scientific community. In other words, an organism’s name is a key to its literature, an entry to what is known about it. Comprehensive accounts on the nomenclature of prokaryotes can be found elsewhere (Bousfield 1993; Sneath 2005), as can practical guidelines for the recognition of new prokaryotic taxa (Trüper 1999, 2005). Once prokaryotes have been rigorously characterized and classified it is a relatively easy matter to name them.
Identification, the final stage of the taxonomic trinity, is sometimes seen as the raison d’etre of prokaryotic systematics due to the importance of accurately identifying unknown organisms, not least pathogenic bacteria. It is both the act and the result of determining whether unknown organisms belong to established and validly named taxa (Krieg 2005). It involves determining the key characteristics of unknown organisms and matching them against databases containing corresponding information on established taxa (Priest 2004). Organisms found to fall outside known groups should be described and classified as new taxa.
Classifications of prokaryotes are not only markedly data dependent but are in a continuous state of development as high quality information becomes available from the application of both new and improved taxonomic methods. Such taxonomies are essentially pragmatic as they are driven by practical imperatives not by theoretical considerations akin to the biological species concept (Goodfellow et al. 1997; Schleifer 2010). Current approaches to the classification of prokaryotes are based upon the integrated use of genotypic and phenotypic features acquired through the application of chemotaxonomic, molecular systematic and numerical and non-numerical phenotypic methods. This practice, known as polyphasic taxonomy, was introduced by Colwell (1970) to signify successive or simultaneous studies on groups of prokaryotes using methods chosen to yield high quality data. The polyphasic approach provides a sound basis for stable nomenclature and reliable identification, essential factors for a practical or utilitarian taxonomy designed to serve diverse end users. Detailed accounts of the polyphasic approach to the classification of prokaryotes are available (Vandamme et al. 1996; Goodfellow et al. 1997; Gillis et al. 2005).
The widespread application of polyphasic taxonomy led to significant improvements in the classification of prokaryotes, notably in groups like the Actinobacteria and Cyanobacteria where traditional approaches based on form and function proved unreliable (Goodfellow and Maldonado 2006; Kroppenstedt and Goodfellow 2006; Gupta 2009). It has not been possible to assemble a recommended set of methods for polyphasic studies as taxonomic toolkits are influenced by the biological properties and ranks of the taxa under study and by the equipment available to investigators. However, sequencing highly conserved macromolecules, notably 16S rRNA genes, has provided valuable data for constructing phylogenies at and above the genus level (Woese 1987; Ludwig and Klenk 2005) whereas DNA:DNA relatedness, molecular fingerprinting and phenotypic techniques are methods of choice for delineating taxa at and below the rank of species (Rosselló-Mora and Amann 2001). It is important to remember that distinguishing phenotypic features are required for the formal description of new species (Wayne et al. 1987). Procedures used to characterize and circumscribe prokaryotic taxa have been considered in detail (Felis et al. 2010; Tindall et al. 2010) and the strengths and weaknesses of genomic methods have been highlighted by Schleifer (2010).
The phylum Actinobacteria
Actinobacterial systematics has been revolutionized by the application of chemotaxonomic, molecular systematic and numerical taxonomic methods (Goodfellow and Cross 1984; Stackebrandt and Schumann 2006). The class Actinobacteria is now seen to be one of the major phyla in the domain Bacteria, as inferred from its branching position in the 16S rRNA gene tree (Ludwig and Klenk 2005). The separation of this taxon from other bacterial groups is supported by conserved indels in protein (e.g. cytochrome-coxidase subunit 1, CTP synthase and glutamyl-tRNA synthase) and 23S rRNA sequences (Gao and Gupta 2005; Gao et al. 2006) and by characteristic gene arrangements (Kunisawa 2007) though it is still not possible to identify the phylogenetically closest neighbours to the actinobacteria with any confidence (Ventura et al. 2007).
The current hierarchical classification of the phylum Actinobacteria is outlined in Fig. 3. The phylogenetic relationships of taxa above the genus level is based solely on taxon-specific 16S rRNA signatures, as spelt out by Zhi et al. (2009) and summarized in Fig. 4. In contrast, classification at generic and species ranks also take into account the discontinuous distribution of chemotaxonomic, morphological and physiological properties, as exemplified by the circumscription of novel genera, such as Actinovallomurus (Tamura et al. 2009) and Plantactinospora (Qin et al. 2009) and by new species of Dactylosporangium (Kim et al. 2010) and Streptomyces (Kumar and Goodfellow 2010). It should be noted that the term actinobacteria refers to all members of the phylum whereas the designation actinomycetes only refers to strains belonging to the order Actinomycetales.

Hierarchic classification of the phylum Actinobacteria

Classification of actinobacteria at the suprageneric level
The current classification of actinobacteria is a marked improvement on earlier taxonomies of the group but needs to be seen as a staging post leading towards better classifications in the future. It can, for instance, be anticipated that the suprageneric relationships between taxa based on 16S rRNA signatures will need to be adjusted as sequences of novel taxa are added to the 16S rRNA Actinobacteria gene tree (Zhi et al. 2009). In addition, the somewhat opaque evolutionary history of the phylum should be clarified by the generation of trees based on whole-genome sequences (Ventura et al. 2007), especially ones taken to represent diverse taxa, not least those lying towards the root of the 16S rRNA tree. It is evident that such a phylogeny-driven approach provides invaluable data for the reconstruction of prokaryotic phylogenetic history and for the discovery of new protein families and biological properties (Wu et al. 2009).
Culture dependent bioprospecting strategy
Marine actinobacteria
In our search for novel natural products that can be developed as resources for healthcare we have focused on the isolation of actinomycetes from extreme and neglected environments, notably deep-sea sediments, on the premise that they are likely to be a rich source of novel strains with the capacity to produce new metabolites (Bull et al. 2005; Maldonado et al. 2005a; Pathom-aree et al. 2006d). The earlier view that actinomycetes in marine habitats were restricted to the genera Micromonospora, Rhodococcus and Streptomyces (Goodfellow and Haynes 1984; Colquhoun et al. 1998) has been comprehensively revised as culture-independent surveys have shown that immense actinobacterial diversity is present in marine habitats (Fenical and Jensen 2006; Gontang et al. 2007; Jensen and Lauro 2008), as exemplified by the estimate of over 1,300 novel actinobacterial taxa in marine sediments based on the application of species richness estimators to microbial diversity data (Stach et al. 2003a, b; Stach and Bull 2005). It is likely that many of these novel taxa will be an important source of new bioactive compounds as they share an evolutionary pedigree with known producers (McVeigh et al. 1996; Ward and Goodfellow 2004; Maldonado et al. 2005a, 2009).
Many cultivable actinomycetes from marine habitats have been characterized and screened in recent times (Fiedler et al. 2005; Fenical and Jensen 2006). Taxonomically diverse communities of actinomycetes have been detected in marine sediments (Jensen et al. 2005a, b; Bredholdt et al. 2007, 2008; Maldonado et al. 2009), notably deep-sea sediments (Pathom-aree et al. 2006d), as well as from mangrove forests (Hong et al. 2009), marine sponges (Kim et al. 2005; Montalvo et al. 2005; Zhang et al. 2006; Jiang et al. 2007), sea cucumbers (Kurahashi et al. 2010) and seaweed (Lee 2008; Lee et al. 2008).
Selective isolation
Our recent studies have been focused on the isolation of actinomycetes from geographically diverse sediment samples using taxon specific isolation procedures. Sediment samples were taken from the Canary Basin (Atlantic Ocean), the Japan Trench (NW Pacific Ocean) and from Norwegian fjords, as described previously (Colquhoun et al. 1998; Stach et al. 2003a, b) and from the deepest place on Earth, the Challenger Deep of the Mariana Trench in the western Pacific Ocean (Pathom-aree et al. 2006d). Isolates were obtained by plating serial dilutions of wet sediment samples, prepared using ¼ strength Ringer’s solution, onto selective isolation media which were incubated at 28°C for 2 or more weeks.
Aliquots of the serial dilutions (75–100 μl) were spread over the surfaces of a range of media known to favour the isolation of specific actinomycete taxa, notably glucose-yeast extract agar supplemented with rifampicin and streptomycin (Athalye et al. 1981), humic acid-vitamin agar (Hayakawa and Nonomura 1987), M3 agar (Rowbotham and Cross 1977), raffinose-histidine agar (Vickers et al. 1984), SM3 agar (Tan et al. 2006) and starch-casein-nitrate agar (Küster and Williams 1964). Agar media were routinely dried for 15 min prior to inoculation, as recommended by Vickers and Williams (1987). All media were supplemented with nystatin (50 μg/ml) to suppress fungal growth.
Many other procedures have been recommended for the selective isolation of actinomycetes from natural ecosystems, including marine habitats (Goodfellow 2010). These include the use of media prepared with natural seawater and the identification of strains that fail to grow when seawater is substituted with deionized water (Jensen et al. 1991; Gontang et al. 2007), which most notably led to the isolation of members of the genus Salinispora (Jensen et al. 1991; Mincer et al. 2002; Maldonado et al. 2005b).
Recognition and dereplication of target actinobacteria
Historically, little attempt was made to establish the effectiveness of selective isolation procedures hence the selection of strains for screening programmes was somewhat arbitrary. This situation, while unsatisfactory, was understandable due to the lack of suitable procedures for the identification of even well established taxa. However, it is now relatively straightforward to determine whether colonies growing on isolation plates belong to target or novel taxa as reliable diagnostic procedures are available for this purpose (The Society for Actinomycetes Japan 2001; Goodfellow et al. 2010a, b).
In general, the assignment of representative isolates growing on isolation plates to target or novel taxa is essentially a two-stage process. Reliable methods are needed to assign isolates to higher taxonomic categories (e.g. genera and families) prior to the selection of diagnostic tests for the recognition of new or validly described species. The assignment of representative isolates to target or putatively novel taxa at and above the genus level can be readily achieved by comparing their full 16S rRNA gene sequences with those of their nearest neighbours drawn from GenBank (for details see Felis et al. 2010) and by evaluating the resultant data in light of appropriate chemotaxonomic and morphological data (The Society for Actinomycetes Japan 2001; Goodfellow et al. 2010a, b). Similarly, the assignment of isolates to established or new species can be achieved by using appropriate combinations of phenotypic tests, as illustrated by the delineation of novel species of Dermacoccus (Pathom-aree et al. 2006b, c), Nonomuraea (Hozzein and Goodfellow 2007), Streptomyces (Khan et al. 2010) and Williamsia (Stach et al. 2004; Pathom-aree et al. 2006a).
The use of taxon-specific oligonucleotide probes as amplification primers offers a practical way of identifying large numbers of target actinomycetes (Mehling et al. 1995; Yoon et al. 1996; Monciardini et al. 2002). Genus-specific probes are available for the one-stop identification of strains of Amycolatopsis (Tan et al. 2006), Gordonia (Shen and Young 2005), Pseudonocardia (Morón et al. 1999) and Saccharomonospora (Salazar et al. 2000), and for members of the Streptomyces violaceusniger 16S rRNA gene clade (Kumar et al. 2007).
Standard diagnostic procedures, such as those outlined above, were used to identify representatives of the different colony types of actinomycetes growing on selective isolation plates seeded with dilutions of the various marine sediment samples. Isolates were placed into genera based on partial or complete 16S rRNA gene sequence data that had been acquired and analysed using appropriate procedures and software packages (Maldonado et al. 2005a; Pathom-aree et al. 2006d). Similarly, selected isolates were assigned to novel or validly described species using combinations of phenotypic criteria, including chemotaxonomic and morphological data (Stach et al. 2004; Pathom-aree et al. 2006a, b, c; Hohmann et al. 2009a; Goodfellow et al. 2010a, b).
Dereplication of isolates
There is a contradiction between formal taxonomic practice and the need to select representatives from extensive actinobacterial populations for screening purposes. Taxonomic studies require a thorough characterization of relatively few isolates whereas the choice of high quality material for pharmacological screens requires the rapid selection of representative strains from many isolates of the target taxa. This tension between the requirements of formal and practical taxonomy was first addressed by Williams et al. (1969).
Williams and his colleagues assigned large numbers of soil streptomycetes to groups based on aerial spore mass, colony reverse and diffusible pigment colours produced on oatmeal agar and on their capacity to form melanin pigments on peptone-yeast extract-iron agar. It was subsequently shown that such colour-groups reflected the extent of the taxonomic diversity of cultivable streptomycetes in rhizosphere and non-rhizosphere soils (Williams and Vickers 1988; Atalan et al. 2000; Sembiring et al. 2000) as isolates taken to represent such taxa key out to either established or novel Streptomyces species or species-groups based on computer-assisted identification (Williams and Vickers 1988; Atalan et al. 2000) and polyphasic taxonomic procedures (Manfio et al. 2003; Goodfellow et al. 2007). Other rapid methods that can be used to rapidly assign actinomycetes to predictive groups include analytical chemical and molecular fingerprinting procedures (Ferguson et al. 1997; Maldonado et al. 2008). Dereplication is the term used for differentiating phenotypically ambiguous strains in order to facilitate efficient screening and thereby minimize costs and time in sorting large collections of isolates (Brandão et al. 2002).
The assignment of streptomycetes to colour-groups has been used to gain an insight into the taxonomic diversity of these organisms in marine sediments (Goodfellow and Haynes 1984; Pathom-aree et al. 2006d) and in a beach and dune sand system (Antony-Babu and Goodfellow 2008) and thereby to the selection of representative isolates for screening assays. We have found that the use of such high quality biological material leads to a marked increase in hit rates, notably from strains isolated from different geographical locations. It is also encouraging that a reasonable linear correlation exists between streptomycete colour- group and corresponding rep-PCR data (Antony-Babu et al. 2010). These workers introduced a computer-assisted numerical system for the objective analysis of colour-group data, and in doing so opened up the prospect of generating cumulative colour-group databases which can be used to objectively select representative streptomycetes for screening.
Novel taxa
To date, members of 50 genera of actinomycetes have been isolated from marine sources (see Table 1). These include isolates assigned to novel genera, including Demequina (Yi et al. 2007), Iamia (Kurahashi et al. 2009), Marinactinospora (Tian et al. 2009a), Marisediminicola (Li et al. 2010), Miniimonas (Ue et al. 2010), Paraoerskovia (Khan et al. 2009), Phycicococcus (Lee 2008), Phycicola (Lee et al. 2008), Salinibacterium (Han et al. 2003), Salinispora (Maldonado et al. 2005b), Sciscionella (Tian et al. 2009b) and Serinicoccus (Yi et al. 2004). In addition, a steady stream of new marine-derived species have been classified in established genera, including Arsenicococcus (Hamada et al. 2009) Dermacoccus (Pathom-aree et al. 2003b, c), Kocuria (Seo et al. 2009), Nocardiopsis (Chen et al. 2009), Saccharomonospora (Liu et al. 2010), Streptomyces (Pimentel-Elardo et al. 2009; Xu et al. 2009; Khan et al. 2010), Williamsia (Stach et al. 2004; Pathom-aree et al. 2006a) and Verrucosispora (Liao et al. 2009; Dai et al. 2010).
Production media
The media used for submerged cultivation of actinomycetes have a dramatic impact on the expression of secondary metabolite gene clusters though, in general, it is not known why. The success or otherwise of screening programmes is not only dependent on the composition of complex media and/or the use of specific carbon and nitrogen sources, but is influenced by the taxonomic status of the organisms under study. A selection of media commonly used by our group for the production of secondary metabolites is shown in Table 2.
Medium 410, which has a high content of both carbon and nitrogen, permits optimal growth of nearly all actinomycetes, notably members of the suborder Corynebacterineae, such as Gordonia, Nocardia, Rhodococcus and Tsukamurella strains. However, this medium is not ideal for inducing secondary metabolite production from actinomycetes which form aerial mycelia (e.g. streptomycetes) or spore vesicles (e.g. streptosporangiae) though it is a good seed medium for the production of biomass from such organisms. In contrast, media 19, 400, OM and SGG promote the production of novel drug candidates from Streptomyces strains whereas members of genera classified in the family Micromonosporaceae are best grown in media 333, MMM and SGG. Consequently, the success of screening programmes for secondary metabolite production are heavily dependent on the assignment of isolates to the correct taxa.
In our experience it is essential to grow dereplicated isolates in a diverse range of production media (Fiedler 1994; Theobald et al. 2000) including the use of formulations which mimic conditions in the environment in the case of strains from marine habitats. However, most of the 700 dereplicated marine isolates assigned to the families Micromonosporaceae, Nocardiaceae, Pseudonocardiaceae and Streptomycetaceae were salt tolerant and did not require seawater for the production of secondary metabolites in submerged culture. Indeed, a few of the isolates showed better growth and enhanced secondary metabolite production in media lacking seawater. The richest and most chemically diverse secondary metabolites detected by HPLC diode array analysis were recorded for members of the genus Streptomyces. Approximately, 26% of extracts from these organisms gave positive results, the corresponding figures for strains assigned to the families Micromonosporaceae, Nocardiaceae and Pseudonocardiaceae were 24, 6.4 and 1.7%.
Monitoring secondary metabolite production
A well established method for analyzing the productivity of strains and the diversity of their secondary metabolite patterns is reversed-phase HPLC in gradient mode coupled with diode array monitoring (Huber and Fiedler 1991). Known metabolites can be excluded by using a database containing many secondary and primary metabolites which were analyzed by using the same HPLC conditions; e.g. our in-house HPLC-UV-Visible absorption spectral database contains more than 950 entries, mainly of antibiotics (Fiedler 1993). Presumptive new metabolites can be characterized by this method according to their UV–Visible properties and retention times. The presumptive novelty of metabolites are then confirmed by HPLC-MS analysis, followed by scale-up fermentation of strains, and isolation and structural elucidation of pure compounds. The application of this screening strategy to freshly isolated strains has resulted in the detection of a high number of identified novel compounds compared to a low throughput of strains (Fiedler 2010). The advantage of this method is based in the separation selectivity and diode array monitoring which permit a very close look at each individual generated extract. The method allows the detection and characterization of broad metabolite patterns in culture filtrates or raw extracts of organisms. However, polar metabolites and metabolites without UV–Visible chromophors cannot be detected in culture broths or extracts by this method.
Alternative methods for the detection of secondary metabolites that are commonly used include target assays based on enzyme or receptor inhibition. Such procedures are preferred by the pharmaceutical industry because of the strong correlation between metabolite, biological activity and target. Nevertheless, such high and ultra-high throughput assays have not led to the marketing of any novel compound to date (Baltz 2005). Small research groups lack the expensive robotic equipment and manpower necessary for such high throughput procedures and hence have to develop individual assays that are easy to handle, as in our detection of abyssomicins, potent and selective inhibitors of the biosynthesis of para-aminobenzoic and folic acids (Riedlinger et al. 2004).
Recent discoveries from our research group
A harmonized collaboration is absolutely necessary for success in research, as can be demonstrated by the discoveries of our broader research team which includes the groups of Professor Roderich D. Süssmuth from the Organic Chemistry Department at the Technical University of Berlin, Professor Marcel Jaspars from the Marine Biodiscovery Centre, Department of Chemistry at the University of Aberdeen, and Professor Alan Bull, School of Biosciences, University of Kent.
Abyssomicins B, C, atrop-C, D, G and H
These unique polycyclic polyketide synthase type 1-antibiotics (Fig. 5) were found using a combination of a target assay and HPLC-diode-array detector (DAD) monitoring. The target, the biosynthesis of para-aminobenzoic acid (Pab), was based on a whole-cell agar plate diffusion assay that permitted the detection of antibiotics which selectively inhibited the biosynthesis of the aromatic amino acids and para-aminobenzoic acid, respectively (Riedlinger et al. 2004). One out of 930 extracts from 201 marine and terrestrial actinomycetes was positive in the assay, an extract generated from Verrucosispora maris AB-18-032 (Goodfellow et al. 2010a, b), which was isolated from a sediment sample collected from the Sea of Japan at a depth of 289 m. HPLC-DAD monitoring of the extract revealed a metabolite family (Fig. 5) in which the main compound, atrop-abyssomicin C, was active against Gram-positive bacteria, including multiresistant—and vancomycin-resistant—Staphylococcus aureus isolates (Riedlinger et al. 2004; Bister et al. 2004; Keller et al. 2007a). All of the abyssomicins mimic the structure of chorismate, the natural substrate for the PabB subunit of 4-amino-4-deoxychorismate synthase, though only abyssomicin C and atrop-abyssomicin C bind covalently to PabB by a Michael addition mechanism (Keller et al. 2007b).

Structures of abyssomicins isolated from Verrucosispora maris AB-18-032
Albidopyrone
This new α-pyrone containing secondary metabolite was detected by HPLC-DAD analysis in a culture filtrate extract of Streptomyces sp. NTK 227, a strain isolated from an Atlantic Ocean sediment and found to be a member of the Streptomyces albidoflavus 16S rRNA gene clade. Albidopyrone shows a moderate inhibitory activity against protein-tyrosine phosphatase B (Hohmann et al. 2009a). The structure of this compound is shown in Fig. 6.

Structure of albidopyrone isolated from Streptomyces sp. NTK 227
Benzoxazine NTK 935
Streptomyces sp. NTK 935 was isolated from an Atlantic Ocean sediment core (3, 814 m) at the southern edge of the Canary Basin. HPLC extracts of the organism showed that it produced a new benzoxazine compound which had a strong inhibitory activity against the enzyme glycogen synthase kinase 3-beta (H-P Fiedler, M Goodfellow, RD Süssmuth, JF Imhoff, unpubl.). The structure of this compound is shown in Fig. 7.

Structure of benzoxazine isolated from Streptomyces sp. NTK 935
Caboxamycin
This new benzoxazole antibiotic was detected by HPLC-diode array screening in extracts of Streptomyces sp. NTK 937, another strain which was isolated from sediment collected from the Canary Basin. The compound, caboxamycin, was named after the first letters of the collection site from which the organism was isolated and from letters drawn from its chemical structure. Caboxamycin showed inhibitory activity against both Gram-positive bacteria and against the tumour cell lines gastric adenocarcinoma (AGS), hepatocellular carcinoma (Hep G2) and breast carcinoma cells (MCF7). The antibiotic also showed an inhibitory activity against the enzyme phosphodiesterase (Hohmann et al. 2009b). Its structure is shown in Fig. 8.

Structure of caboxamycin isolated from Streptomyces sp. NTK 937
Dermacozines
Nineteen out of 38 actinomycetes isolated from a sediment sample collected from the Challenger Deep (10898 m) of the Mariana Trench, using the remotely operated submersible, Kaiko, were found to belong to the genus Dermacoccus (Pathom-aree et al. 2006d). HPLC-DAD analysis of culture filtrates of these isolates showed an interesting pattern of secondary metabolites which were considered to be a group of phenazine compounds. High-resolution mass spectrometry and structural eludication of the compounds carried out by the group of Professor Marcel Jaspars at the University of Aberdeen resulted in the identification of 14 novel phenazine-type metabolites which were named dermacozines. The structure of seven of these compounds have been determined (Fig. 9), they show antitumour, antiprotozoal and free radical scavenging activities (Abdel-Mageed et al. 2010).

Structures of dermacozines extracted from Dermacoccus sp. MT1.1 and MT1.2 which were isolated from the Challenger Deep of the Mariana Trench
Lipocarbazoles A1–A4
A family of new secondary metabolites with a carbazole moiety and an alkyl side chain were detected by HPLC-DAD analysis in cell extracts of Tsukamurella pseudospumae strain Acta 1857, an organism isolated from activated sludge foam collected at Stoke Bardolph Water Reclamation Works, near Nottingham, UK. The metabolites, which were named lipocarbazoles in accordance with their chemical structure, exhibited strong free radical scavenging activity (Schneider et al. 2009). Interestingly, the same secondary metabolite pattern was detected in Tsukamurella strains isolated from the sediment collected from the Challenger Deep of the Mariana Trench (M Goodfellow, AT Bull, H-P Fiedler, unpubl.). The structures of these new metabolites are shown in Fig. 10.

Structures of lipocarbazoles isolated from Tsukamurella pseudospumae Acta 1857 andf rom deep-sea Tsukamurella strains
Lysolipin
Streptomyces sp. NTK 963 was isolated from the same Canary Basin deep-sea sediment sample as the caboxamycin producer, Streptomyces sp. NTK937. HPLC-DAD and HPLC-MS analysis of extracts of this organism showed that it produced lysolipin, an interesting antibiotic with strong antitumour and antibacterial activity. This compound was initially detected in Streptomyces violaceusniger Tü 96, a strain isolated from soil collected in Ajhu, India (Drautz et al. 1975) and re-discovered in Streptomyces tendae Tü 4042, an isolate from an arid soil sample collected near Alice Springs in Australia. The structure of this compound is shown in Fig. 11.

Structure of lysolipin I isolated from Streptomyces sp. NTK 963
Proximicins A, B and C
These novel aminofuran-type antibiotics were detected by HPLC-DAD analysis in extracts of Verrucosispora strain MG-37, an organism which was isolated from a sediment sample collected from the Raune Fjord, Norway at a depth of 250 m. One of these compounds, proximicin A, was detected in the abyssomicin producer Verrucosispora maris AB-18-032 (Fiedler et al. 2008). The characteristic structural element of proximicins is 4-amino-furan-2-carboxylic acid, a hitherto unknown γ-amino acid (Schneider et al. 2008). Proximicins exhibit weak antibacterial activity but have a strong cytostatic effect against various human tumour cell lines. All of the proximicins showed significant growth inhibitory activity towards gastric adenocarcinoma (AGS) and hepatocellular carcinoma (Hep G2) though breast carcinoma cells (MCF 7) were less sensitive. The proximicins were found to arrest AGS cells in the G0/G1 phase of the cell-cycle and increase the level of cell-cycle regulatory proteins p53 and p21 (Schneider et al. 2008). The structures of the proximicins are shown in Fig. 12.

Structures of proximicins isolated from Verrucosispora sp. MG-37
Conclusions
Overview and new directions of travel
It can be concluded from the application of our bioprospecting strategy that a combination of selective isolation, strain dereplication and screening procedures can lead to the discovery of new natural products from novel actinomycetes isolated from geographically diverse sediment samples, as exemplified by the production of atrop-abyssomicin C from V. maris (Bister et al. 2004; Riedlinger et al. 2004; Keller et al. 2007a, b; Goodfellow et al. 2010a, b), the dermacozines from Dermacoccus species (Abdel-Mageed et al. 2010) and caboxamycin and proximcins A, B and C from putatively novel species of Streptomyces and Verrucosispora, respectively (Schneider et al. 2008; Hohmann et al. 2009b). These results provide further evidence that marine-derived actinomycetes are an important source of new secondary metabolites (Magarvey et al. 2004; Fiedler et al. 2005; Fenical and Jensen 2006; Lam 2006; Bull and Stach 2007; Williams 2008; Olano et al. 2009a, b).
Our results help underpin the re-emerging concept that taxonomic diversity can be used as a surrogate for chemical diversity amongst actinomycetes, especially at the species level (Ward and Goodfellow 2004; Goodfellow et al. 2007; Tan et al. 2007). The strongest evidence for this concept comes from extensive studies on the genus Salinispora, a taxon which encompasses two valided described species, Salinispora arenicola and S. tropica (Maldonado et al. 2005b) and the presumptive new species, ‘Salinispora pacifica’ (Fenical and Jensen 2006). These bacteria are widely distributed in marine sediments (Jensen and Mafnas 2006) and are a rich source of structurally unique secondary metabolites (Feling et al. 2003; Fenical and Jensen 2006; Williams et al. 2007; Oh et al. 2008; Asolkar et al. 2009), including salinosporamide A which is presently in clinical trials for the treatment of cancer (Fenical et al. 2009). The three Salinispora species synthesize a range of species-specific metabolites; S. arenicola strains produce rifamicin derivatives and staurosporine analogues and S. tropica strains salinosporamides and sporalides (Fenical and Jensen 2006; Jensen 2010). In contrast, some ‘S. pacifica’ strains produce the structurally novel metabolites cyanosporasides A and B and others the polyketides pacificanones A and B, and salinipyrones A and B (Oh et al. 2008). These findings chime with reports that the secondary metabolite profiles of filamentous fungi are species-specific (Larsen et al. 2005; Frisvad et al. 2008; Frisvad 2010).
The couplings between taxonomic and chemical diversity are at variance with the widely held view that secondary metabolite production is strain specific, a stance partly based on the contention that Streptomyces species, notably Streptomyces griseus and Streptomyces hygroscopicus, synthesize diverse secondary metabolites and on the proposition that taxonomy is not necessarily a good indicator of bioactive potential (Strohl 2004). However, claims that S. griseus and S. hygroscopicus encompasses strains with diverse secondary metabolite profiles need to be re-assessed in light of the improved taxonomy of these species (Guo et al. 2008; Kumar and Goodfellow 2008), especially since several new Streptomyces species are based on strains previously misclassified as S. hygroscopicus (Kumar and Goodfellow 2010). Indeed, it would be interesting to see if coupling between taxonomic and chemical diversity occurs below the species level, a hypothesis that could be tested by screening S. griseus ecovars isolated from sampling sites taken along a transect across a beach and dune-sand system (Antony-Babu et al. 2008). Representatives of this species would be ideal candidates for testing this proposition as bona fide members of this taxon are a source of new bioactive compounds (Piel 2004; Graf et al. 2007).
The proposition that the search for new actinomycete diversity is an important element in our drug discovery strategy (Fig. 2) is strongly supported by comparative full genome sequence data of the type strains of S. arenicola and S. tropica (Penn et al. 2009; Jensen 2010). A comparison of the full genome sequences of these strains helps explain the ability of members of the two species to produce core sets of species-specific secondary metabolites and allows insight into the processes that drive speciation in this genus. The most interesting feature drawn from a comparison of the two genomes is that species-specific genes are concentrated in genomic islands (Coleman et al. 2006; Penn et al. 2009). These islands are sites within which niche specific genes, including the biosynthetic genes linked to Salinispora species-specific secondary metabolite production, are located and ecological adaptation between the three closely related Salinispora species resolved. The species-specificity of Salinispora biosynthetic pathways (Jensen et al. 2007) strongly supports the view that secondary metabolites can provide valuable taxonomic information, as inferred from studies on Amycolatopis regifaucium (Tan et al. 2007), Streptomyces clavuligerus (Ward and Goodfellow 2004) and the Streptomyces violaceusniger 16S rRNA gene clade (Goodfellow et al. 2007).
Isolates representing a broad range of taxa are needed to assess the chemical and genetic diversity of marine actinobacteria and hence their full potential as a source of novel secondary metabolites. However, representatives of relatively few taxa have been isolated from marine as opposed to terrestrial habitats (see Goodfellow 2010). In general, many of the media formulations used to isolate actinomycetes from marine sources have been somewhat empirical and have led to the isolation of relatively small numbers of strains belonging to a few established taxa (Jiang et al. 2007; Bredholdt et al. 2007, 2008; Hong et al. 2009; Maldonado et al. 2009). In contrast, large numbers of strains have been isolated using reliable selective isolation procedures, notably ones used to isolate members of the genera Micromonospora (Maldonado et al. 2008; Qui et al. 2008), Rhodococcus (Colquhoun et al. 1998) and Streptomyces (Goodfellow and Haynes 1984; Jensen et al. 1991), as well as the seawater requiring genus Salinispora (Mincer et al. 2002; Jensen et al. 2005a, b).
The application of additional selective isolation methods can be expected to yield additional taxonomic diversity from the marine biome, such as procedures which have shown that members of the genera Actinomadura (Athalye et al. 1981), Amycolatopsis (Tan et al. 2006), Planobispora (Suzuki et al. 2001b) and Planomonospora (Suzuki et al. 2001a) are common and widespread in terrestrial habitats. In addition, new selective procedures are available for the isolation of alkaliphilic streptomycetes (Antony-Babu and Goodfellow 2008) and for members of the Streptomyces violaceoruber 16S rRNA gene clade (Duangmal et al. 2005).
It will also be necessary to devise innovative selective isolation strategies to isolate novel actinobacteria detected in culture-independent surveys of marine habitats (Stach et al. 2003a, b; Kim et al. 2004; Gontang et al. 2007; Jensen and Lauro 2008) and additional strains of genera such as Verrucosispora which are potential sources of novel secondary metabolites. However, there is an even more urgent requirement to focus on the isolation of understudied taxa, such as members of the families Conexibacteriaceae, Coriobacteriaceae and Rubrobacteriaceae, which form deep lineages in the 16S rRNA actinobacterial tree (Zhi et al. 2009) even though members of these taxa isolated from terrestrial sources have not been shown to be prolific sources of bioactive compounds. The isolation of members of such taxa may require leads from bioinformatic analyses of representative whole-genome sequences. Other methods which might be used to good effect include the use of long incubation times (Salt et al. 2002; Gontang et al. 2007), in situ procedures (Epstein et al. 2010), dilution to extinction culturing (Stingl et al. 2008) and cultivation approaches using electron acceptors and substrate gradients (Kopke et al. 2005).
Additional thought needs to be given to the selection of marine habitats for study, to representative sampling, and to the ecology of target organisms (Bull et al. 2005; Bull and Stach 2007; Goodfellow 2010; Jensen 2010). Taxonomic landscapes of marine actinomycetes generated by terminal restriction fragment—length RFLP (T-RFLP) or by single-strand conformation polymorphism (SSCP) of clone libraries or by analysis of community 16S rRNA can be used to select appropriate sampling sites (Stach et al. 2003a; Bull et al. 2005; Maldonado et al. 2005a). Physico-chemical interactions between microorganisms and particulate matter influence the composition of inocula. The dispersion and differential centrifugation technique (DDC), a multistage procedure introduced by Hopkins et al. (1991), combines several physico-chemical treatments which are effective in increasing the yield and diversity of actinobacteria from natural habitats (MacNaughton and O’Donnell 1994; Atalan et al. 2000; Sembiring et al. 2000), including marine sediments (Mexson 2000; Maldonado et al. 2005b). The DDC procedure, for instance, yielded fivefold increases in actinobacteria isolated from a fjord sediment (Maldonado et al. 2005b) and has led to the delineation of several new Streptomyces species, albeit from soil (Manfio et al. 2003; Goodfellow et al. 2007). In addition, the seminal studies on Salinispora show that increased efforts are needed to isolate novel actinobacteria with an obligate requirement for sodium. Studies on the growth and metabolic activities of actinobacteria in situ might help to inform approaches to targeting the isolation of a greater diversity of indigenous marine actinobacteria; ecophysiological approaches have led to the isolation of novel actinobacteria from activated sludge systems (Seviour et al. 2008).
It can be anticipated that technological change, especially in bioinformatics, genomics, metagenomics and metabolite profiling, will greatly influence approaches to the selective isolation, dereplication and characterization and hence on the selection of novel marine actinobacteria for screens (Bull and Stach 2007; Wu et al. 2009; Jensen 2010). Phylogenetic analyses of biosynthetic genes, for instance, are already fostering new methods for predicting secondary metabolite production thereby maximizing opportunities for drug discovery. Strain selection will be critical for such studies so there will be a requirement for improved dereplication technologies, notably the use of molecular screens to rapidly highlight isolates with the greatest genetic potential to produce both target and new secondary metabolites.
Systematics in the post-genomic era
Actinobacterial systematics has played a significant role in the discovery of secondary metabolites from novel actinomycetes isolated from marine habitats. These advances were made at a time when microbial systematics was being seen to be in a state of critical decline (House of Lords Science and Technology Committee 2008) and when the use of strain names was vying with the practice of employing formal species names underpinned by the International Code of Nomenclature of Bacteria (Lapage et al. 1975, 1992). Type strains not only provide anchor points for the names of prokaryotic species but their full 16S rRNA gene sequences are essential for comparative purposes now that sequences of this gene are the primary means used in the initial taxonomic assignments of putative novel isolates. A drift towards the abandonment of the nomenclatural type concept risks a return to the pre-Bergey days of classification where many common bacteria carried a confusing mix of multiple names and where strains were so poorly described that they were difficult to tell apart.
Nevertheless, the current classification of prokaryotes, as exemplified in the present edition of Bergey’s Manual of Systematic Bacteriology (De Vos et al. 2009; Goodfellow et al. 2010a, b), is based on a utilitarian model, the polyphasic approach, which draws upon genotypic and phenotypic data, as well as phylogenetic information (Vandamme et al. 1996; Goodfellow et al. 1997; Schleifer 2010). The extensive application of polyphasic taxonomy has led to revolutionary improvements in prokaryotic systematics, including the classification of actinobacteria (Goodfellow et al. 2010a, b). Indeed, this approach has helped clarify relationships between even the most closely related Streptomyces species (Goodfellow et al. 2007; Kumar and Goodfellow 2008, 2010). In addition, major differences can be found in actinobacterial 16S rRNA gene trees generated by different workers (e.g. Ludwig and Klenk 2005; Zhi et al. 2009). These result from different strategies chosen to construct the gene trees and different sequences used for the analyses. There is no sound basis to accept one tree over another, and experience has shown that some ambiguities remain in most phylogenetic trees for any gene, including the 16S rRNA gene.
Schleifer (2010) has reminded us that the ultimate aim is to generate a theory-based classification grounded on a phylogenetic/evolutionary concept. He also made it clear that even in light of genomic fluidity there is a strong case for believing that the typical genotypic and phenotypic characteristics of taxa are maintained and sufficient for reliable classification and identification, as exemplified by the maintenance of the species-specific properties of the three species of Salinispora (Jensen 2010). There is also a wealth of evidence which shows that well-defined genotypic clusters are congruent with known species circumscribed using polyphasic approaches (Konstantinidis and Tiedje 2005). There are, therefore, good grounds for continuing with and extending current polyphasic classifications while the merits of theory-based alternatives are explored (Staley 2006; Konstantinidis and Tiedje 2007; Achtman and Wagner 2008; Koeppel et al. 2008). There is, however, a real need to find quicker and more reliable procedures for describing new species as current methods are laborious and time-consuming.
It can be concluded that advances in prokaryotic systematics have provided well-defined taxa, a stable nomenclature and improved identification systems which have contributed to developments in actinobacterial biology, not least in strategies for natural product discovery. A lot has been achieved but much remains to be done. There is now an urgent need to train, support and employ the next generation of actinobacterial systematists, a process that needs to be addressed by the microbiological community as it cannot solely be left to the vagaries of ‘market forces’.
References
Abdel-Mageed WM, Milne BF, Wagner M, Schumacher M, Sandor P, Pathom-aree W, Goodfellow, Bull AT, Horikoshi K, Ebel R, Diedrich M, Fiedler H-P, Jaspars M (2010) Dermacozines, a new phenazine family from deep-sea dermacocci isolated from a Mariana Trench sediment. Org Biomol Chem 8:2352–2362
Achtman M, Wagner M (2008) Microbial diversity and the genetic nature of microbial species. Nat Rev Microbiol 6:431–440
Allgaier M, Grossart H-P (2006) Diversity and seasonal dynamics of Actinobacteria populations in four lakes in Northeastern Germany. Appl Environ Microbiol 72:3489–3497
Antony-Babu S, Goodfellow M (2008) Biosystematics of alkaliphilic streptomycetes isolated from seven locations across a beach and dune sand system. Antonie van Leeuwenhoek 94:581–591
Antony-Babu S, Stach JEM, Goodfellow M (2008) Genetic and phenotypic evidence for Streptomyces griseus ecovars isolated from a beach and dune sand system. Antonie van Leeeuwenhoek 94:63–74
Antony-Babu S, Stach JEM, Goodfellow M (2010) Computer-assisted numerical analysis of colour group data for dereplication of streptomycetes for bioprospecting and ecological purposes. Antonie van Leeuwenhoek 97:231–239
Asolkar RN, Freel KC, Jensen PR, Fenical W, Kondratyuk ParkE-J, Pezzuto JM (2009) Arenamides A-C, cytotoxic NFkB inhibitors from the marine actinomycete Salinispora arenicola. J Nat Prod 72:396–402
Atalan E, Manfio GP, Ward AC, Kroppenstedt RM, Goodfellow M (2000) Biosystematic studies on novel streptomycetes from soil. Antonie van Leeuwenhoek 77:337–353
Athalye M, Lacey J, Goodfellow M (1981) Selective isolation and enumeration of actinomycetes using rifampicin. J Appl Microbiol 51:289–291
Baltz RH (2005) Antibiotic discovery from actinomycetes: will a renaissance follow the decline and fall? SIM News 55:186–196
Baltz RH (2008) Renaissance in antibacterial discovery from actinomycetes. Curr Opin Pharmacol 8:557–563
Bentley SD, Cerdano-Tarraga AM, Challis GL et al (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147
Bérdy J (2005) Bioactive microbial metabolites. J Antibiot 58:1–26
Bister B, Bischoff D, Ströbele M, Riedlinger J, Reicke A, Wolter F, Bull AT, Zähner H, Fiedler H-P, Süssmuth RD (2004) Abyssomicin C—a polycyclic antibiotic from a marine Verrucosispora strain as an inhibitor of the p-aminobenzoic acid/tetrahydrofolate biosynthesis pathway. Angew Chem Int Ed 43:2574–2576
Bousfield IJ (1993) Bacterial nomenclature and its role in systematics. In: Goodfellow M, O’Donnell AG (eds) Handbook of new bacterial systematics. Academic Press, London, pp 317–338
Brandão PFB, Torimura M, Kurane R, Bull AT (2002) Dereplication for biotechnology screening: PyMS and PCR-RFLP-SSCP (PRS) profiling of 16S rRNA genes of marine and terrestrial actinomycetes. Appl Microbiol Biotechnol 58:77–83
Bredholdt H, Galatenko OA, Engelhardt K, Tjaervik E, Terekhova LP, Zotchev SB (2007) Rare actinomycete bacteria from the shallow water sediments of the Trondheim fjord, Norway: isolation, diversity and biological activity. Environ Microbiol 9:2756–2764
Bredholdt H, Tjaervik E, Johnsen G, Zotchev SB (2008) Actinomycetes from sediments in the Trondheim fjord, Norway: diversity and biological activity. Mar Drugs 6:12–24
Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds) (2005) Bergey’s manual of systematic bacteriology, 2nd edn, vol 2, part A, introductory essays. Springer, USA, pp 1–304
Bull AT (2004a) Biotechnology, the art of exploiting biology. In: Bull AT (ed) Microbial diversity and bioprospecting. ASM Press, Washington, pp 3–12
Bull AT (2004b) The paradigm shift in microbial prospecting. In: Bull AT (ed) Microbial diversity and bioprospecting. ASM Press, Washington, pp 241–249
Bull AT (2010) Actinobacteria of the extremobiosphere. In: Horikoshi K, Antranikian G, Bull AT, Robb F, Stelter K (eds) Extremophiles handbook. Springer-Verlag GmbH, Berlin (in press)
Bull AT, Stach JEM (2007) Marine actinobacteria: new opportunities for natural product search and discovery. Trends Microbiol 15:491–499
Bull AT, Ward AC, Goodfellow M (2000) Search and discovery strategies for biotechnology: the paradigm shift. Microbiol Mol Biol Rev 64:573–606
Bull AT, Stach JEM, Ward AC, Goodfellow M (2005) Marine actinobacteria; perspectives, challenges, future directions. Antonie van Leeuwenhoek 87:65–79
Chen Y-G, Wang Y-X, Zhang Y-Q, Tang S-K, Liu Z-X, Xioa H-D, Xu L-H, Cui X-L, Li W-J (2009) Nocardiopsis litoralis sp. nov., a halophilic marine actinomycete isolated from a sea anemone. Int J Syst Evol Microbiol 59:2708–2713
Coleman ML, Sullivan MB, Martiny AC, Barry K, Delong EF, Chisholm SW (2006) Genomic islands and the ecology and evolution of Prochlorococcus. Science 311:1768–1770
Colquhoun JA, Mexson J, Goodfellow M, Ward AC, Horikoshi K, Bull AT (1998) Novel rhodococci and other mycolate actinomycetes from the deep sea. Antonie van Leeuwenhoek 74:27–40
Colwell RR (1970) Polyphasic taxonomy of bacteria. In: Iizaka H, Hasegawa T (eds) Culture collections of microorganisms. University of Tokyo Press, Tokyo, pp 421–436
Czaran TL, Hoekstra RE, Page L (2002) Chemical warfare between microbes promotes biodiversity. Proc Natl Acad Sci USA 99:786–790
Dai H-Q, Wang J, Xim Y-H, Pei G, Tang S-K, Ren B, Ward A, Ruan J-S, Li W-J, Zhang L-X (2010). Verrucosispora sediminis sp. nov., a novel cyclodipeptide producing actinomycete from the South China Sea. Int J Syst Evol Microbiol. doi:10.1099/ijs.0.017053-0
De Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer K-H, Whitman WB (eds) (2009) Bergey’s manual of systematic bacteriology, 2nd edn, vol 3, The Firmicutes. Springer, USA, pp 1–1450
Drautz H, Keller-Schierlein W, Zähner H (1975) Lysolipin I, ein neues Antibiotikum aus Streptomyces violaceoniger. Arch Microbiol 106:175–190
Duangmal K, Ward AC, Goodfellow M (2005) Selective isolation of members of the Streptomyces violaceoruber clade from soil. FEMS Microbiol Lett 243:321–327
Epstein, Lewis K, Nichols D, Gavrish E (2010) New approaches to microbial isolation. In: Manual of industrial microbiology and biotechnology, 3rd edn. Baltz RH, Davies J, Demain AL (volume eds) Section 1: Isolation and screening for secondary metabolites and enzymes, Bull AT, Davies JE (section eds). ASM Press, Washington, DC, pp 3–12
Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W (2003) Salinosporamide A: a high cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew Chem Int Ed 42:355–357
Felis GE, Torriani S, van Hylckama Vlieg JT, Oren A (2010) Taxonomic characterization of prokaryotic microorganisms. In: Manual of industrial microbiology and biotechnology, 3rd edn. Baltz RH, Davies J, Dermain AL (volume eds) Section 1: Isolation and screening of secondary metabolites and enzymes, Bull AT, Davies J (section eds). ASM Press, Washington, DC, pp 28–42
Fenical W, Jensen PR (2006) Developing a new resource for drug discovery: marine actinomycete bacteria. Nat Chem Biol 2:666–673
Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC (2009) Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Biorg Med Chem 17:2175–2180
Ferguson EV, Ward AC, Sanglier J-J, Goodfellow M (1997) Evaluation of Streptomyces species-groups by pyrolysis mass spectrometry. Zbl Bakt 285:169–181
Fiedler H-P (1993) Biosynthetic capacities of actinomycetes. 1. Screening for secondary metabolites by HPLC and UV-visible absorbance spectral libraries. Nat Prod Lett 2:119–128
Fiedler H-P (1994) Biosynthetic capacities of actinomycetes. 2. Juglomycin Z, a new naphthoquinone antibiotic from Streptomyces tendae. J Antibiot 47:1116–1122
Fiedler H-P (2010) Secondary metabolites isolated by the Fiedler group. http://www.mikrobio.uni-tuebingen.de/ag_fiedler/index.php
Fiedler H-P, Bruntner C, Bull AT, Ward AC, Goodfellow M, Potterat O, Puder C, Mihm M (2005) Marine actinomycetes as a source of novel secondary metabolites. Antonie van Leeuwenhoek 87:37–42
Fiedler H-P, Bruntner C, Riedlinger J, Bull AT, Knutsen G, Goodfellow M, Jones AL, Maldonado L, Pathom-aree W, Beil W, Schneider K, Keller S, Süssmuth RD (2008) Proximicin A, B and C, novel aminofuran antibiotic and anticancer compounds isolated from marine strains of the actinomycete Verrucosispora. J Antibiot 61:158–163
Frisvad JC (2010) Metabolonics for the discovery of novel compounds. In: Manual of industrial microbiology and biotechnology, 3rd edn. Baltz RH, Davies J, Demain AL (volume eds) Section 1: Isolation and screening of secondary metabolites and enzymes, Bull AT, Davies JE (section eds). ASM Press, Washington, DC, pp 73–77
Frisvad JC, Andersen B, Thrane U (2008) The use of secondary metabolite profiling in chemotaxonomy of filamentous fungi. Mycol Res 112:231–240
Fry JS (2004) Culture dependent microbiology. In: Bull AT (ed) Microbial diversity and bioprospecting. ASM Press, Washington, DC, pp 80–91
Gao B, Gupta RS (2005) Conserved indels in protein sequences that are characteristic of the phylum Actinobacteria. Int J Syst Evol Microbiol 55:2401–2412
Gao B, Paramanathan B, Gupta RS (2006) Signature proteins that are characteristic of Actinobacteria and their subgroups. Antonie van Leeuwenhoek 90:69–91
Gillis M, Vandamme P, De Vos P, Swings J, Kersters K (2005) Polyphasic taxonomy. In: Brenner DJ, Krieg R, Staley JT, Garrity GM (eds) Bergey’s manual of systematic bacteriology, 2nd edn, vol 2, the proteobacteria, part A, introductory essays. Springer, USA, pp 43–48
Giovanonni SJ, Stingl U (2005) Molecular diversity and ecology of microbial plankton. Nature 437:343–348
Gontang EA, Fenical W, Jensen PR (2007) Phylogenetic diversity of Gram-positive bacteria cultured from marine sediments. Appl Environ Microbiol 73:3272–3282
Goodfellow M (2010) Selective isolation of actinobacteria. In: Manual of industrial microbiology and biotechnology, 3rd edn. Baltz RH, Davies J, Demain AL (volume eds) Section 1: Isolation and screening of secondary metabolites and enzymes, Bull AT, Davies JE (section eds). ASM Press, Washington, DC, pp 13–27
Goodfellow M, Cross T (1984) Classification. In: Goodfellow M, Mordarski M, Williams ST (eds) The biology of the actinomycetes. Academic Press, London, pp 7–164
Goodfellow M, Haynes JA (1984) Actinomycetes in marine sediments. In: Ortiz-Ortiz L, Bojalil LF, Yakoleff V (eds) Biological, biochemical and biomedical aspects of actinomycetes. Academic Press, Orlando, pp 453–472
Goodfellow M, Maldonado LA (2006) The families Dietziaceae, Gordoniaceae, Nocardiaceae and Tsukamurellaceae. In: Dworkin M, Falkow S, Schleifer KH, Stackebrandt E (eds) The prokaryotes, 3rd edn, vol 3, Archaea and Bacteria: Firmicutes, Actinomycetes. Springer, New York, pp 843–888
Goodfellow M, Manfio GP, Chun J (1997) Towards a practical species concept for cultivable bacteria. In: Claridge MF, Dawah HA, Wilson MR (eds) Species, the units of diversity. Chapman and Hall, London, pp 25–59
Goodfellow M, Kumar Y, Labeda DP, Sembiring L (2007) The Streptomyces violaceusniger clade: a home for streptomycetes with rugose ornamented spores. Antonie van Leeuwenhoek 92:173–199
Goodfellow M, Maldonado LA, Jones AL, Mexson J, Fiedler H-P, Stach JEM, Bull AT (2010a) Verrucosispora maris sp. nov., a novel actinomycete isolated from a marine sediment which produces abyssomicins. Int J Syst Evol Microbiol (in press)
Goodfellow M, Kämpfer P, Busse HJ, Trujillo M, Suzuki K-E, Ludwig W, Whitman WB (2010b) Bergey’s manual of systematic bacteriology, 2nd edn, vol 3, The Actinobacteria. Springer, USA (in press)
Graf E, Schneider K, Nicholson G, Ströbele M, Jones AL, Goodfellow M, Beil W, Süssmuth D, Fiedler HP (2007) Elloxazinones A and B, new aminophenoxazinones from Streptomyces griseus ACTA 2871. J Antibiot 60:277–284
Guo Y, Zhang W, Rong X, Huang Y (2008) A multilocus phylogeny of the Streptomyces griseus 16S rRNA gene clade: use of multilocus sequence analysis for streptomycete systematics. Int J Syst Evol Microbiol 58:149–159
Gupta RS (2009) Protein signatures (molecular synapomorphies) that are distinctive characteristics of the major cyanobacterial clades. Int J Syst Ecol Microbiol 59:2510–2526
Hahn MW (2009) Description of seven candidate species affiliated with the phylum Actinobacteria, representing planktonic freshwater bacteria. Int J Syst Evol Microbiol 59:112–117
Hamada M, Iino T, Iwami T, Tamura T, Harayama S, Suzuki K-I (2009) Arsenicococcus piscis sp. nov., a mesophilic actinobacterium isolated from the intestinal tract of a fish. Actinomycetologia 23:40–45
Han SK, Nedashkviskaya OI, Mikhailov VV, Kim SB, Bae KS (2003) Salinibacterium amurskyyense gen. nov., sp. nov., a novel genus of the family Microbacteriaceae from the marine environment. Int J Syst Evol Microbiol 53:2061–2066
Handelsman J (2004) Soils—the metagenomics approach. In: Bull AT (ed) Microbial diversity and bioprospecting. ASM Press, Washington, pp 109–119
Hayakawa M, Nonomura H (1987) Humic acid-vitamins agar, a new medium for the selective isolation of soil actinomycetes. J Ferment Technol 65:501–509
Hohmann C, Schneider K, Bruntner C, Brown R, Jones AL, Goodfellow M, Kramer M, Imhoff JF, Nicholson G, Fiedler H-P, Süssmuth RD (2009a) Albidopyrone, a new α-pyrone-containing metabolite from marine-derived Streptomyces sp. NTK 227. J Antibiot 62:75–79
Hohmann C, Schneider K, Bruntner C, Irran E, Nicholson G, Bull AT, Jones AL, Brown R, Stach JEM, Goodfellow M, Beil W, Krämer M, Imhoff JF, Süssmuth RD, Fiedler H-P (2009b) Caboxamycin, a new antibiotic of the benzoxazole family and phosphodiesterase inhibitor, produced by the deep-sea strain Streptomyces sp. NTK 937. J Antibiot 62:99–104
Hong K, Gao A-H, Xie Q-Y, Gao H, Zhuang L, Lin H-P, Yu H-P, Yao X-S, Goodfellow M, Ruan J-S (2009) Actinomycetes for marine drug discovery isolated from mangrove soils and plants in China. Mar Drugs 7:24–44
Hopkins DW, MacNaughton SJ, O’Donnell AG (1991) A dispersion and differential centrifugation technique for representative sampling microorganisms from soil. Soil Biol Biochem 23:217–225
House of Lords, Science and Technology Committee 5th Report (2007–2008) Systematics and taxonomy: follow-up. The Stationary Office Limited, London, pp 1–330
Hozzein WN, Goodfellow M (2007) Nonomuraea aegyptia sp. nov., a novel actinomycete isolated from a sand dune. Antonie van Leeuwenhoek 92:165–171
Huber L, Fiedler H-P (1991) HPLC with computerized diode array detection in pharmaceutical research. In: Fong GW, Lam SK (eds) HPLC in the pharmaceutical industry. Marcel Dekker, New York, pp 123–146
Ikeda H, Ishikawa J, Hanamoto A, Shinose H, Kikuchi T, Shiba Y, Sakoki Y, Hattori M, Ōmura S (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol 21:526–531
Jensen PR (2010) Linking species concepts to natural product discovery in the post-genomic era. J Ind Microbiol Biotechnol 37:219–224
Jensen PR, Lauro FM (2008) An assessment of actinobacterial diversity in the marine environment. Antonie van Leeuwenhoek 94:51–62
Jensen PR, Mafnas C (2006) Biogeography of the marine actinomycete Salinispora. Environ Microbiol 8:1881–1888
Jensen PR, Dwight R, Fenical W (1991) Distribution of actinomycetes in near-shore tropical marine sediments. Appl Microbiol 57:1102–1108
Jensen PR, Gontang E, Mafnas C, Mincer TJ, Fenical W (2005a) Culturable marine actinomycete diversity from tropical Pacific Ocean sediments. Environ Microbiol 7:1039–1048
Jensen PR, Mincer TJ, Williams PG, Fenical W (2005b) Marine actinomycete diversity and natural product discovery. Antonie van Leeuwenhoek 87:43–48
Jensen PR, Williams PG, Oh CD, Zeigker L, Fenical W (2007) Species specific secondary metabolite production in marine actinomycetes of the genus Salinispora. Appl Environ Microbiol 73:1146–1152
Jiang S, Sun W, Chen M, Dai S, Zhang L, Liu Y, Lee KJ, Li X (2007) Diversity of culturable actinobacteria isolated from marine sponge Haliclona sp. Antonie van Leeuwenhoek 92:405–416
Keller S, Nicholson G, Drahl C, Sorensen E, Fiedler H-P, Süssmuth RD (2007a) Abyssomicins G and H and atrop-abyssomicin C from the marine Verrucosispora strain AB-18-032. J Antibiot 60:391–394
Keller S, Schadt HS, Ortel I, Süssmuth RD (2007b) Action of atrop-abyssomicin C as an inhibitor of 4-amino-4-deoxychorismate synthease PabB. Angew Chem Int Ed 46:8284–8286
Khan ST, Harayama S, Tamura T, Ando K, Takagi M, Kazuo S-Y (2009) Paraoerskovia marina gen. nov., sp. nov., an actinobacterium isolated from marine sediment. Int J Syst Evol Microbiol 59:2094–2098
Khan ST, Tamura T, Takagi M, Shin-Ya K (2010) Streptomyces tatejamensis sp. nov., Streptomyces marinus sp. nov. and Streptomyces haliclonae sp. nov., three novel species of Streptomyces isolated from marine sponge Haliclona sp. Int J Syst Evol Microbiol. doi:10.1099/ijs.0.019869-0
Kim BS, Oh HN, Kang H, Park SS, Chun J (2004) Remarkable bacterial diversity in the tidal flat sediment as revealed by 16S rRNA analysis. J Microbiol Biotechnol 14:205–211
Kim TK, Garson MJ, Fuerst JA (2005) Marine actinomycetes related to the “Salinispora” group from the Great Barrier Reef sponge Pseudoceratina clavata. Environ Microbiol 7:509–519
Kim B-Y, Stach JEM, Weon H-Y, Kwon S-W, Goodfellow M (2010) Three new species of Dactylsporangium isolated from soil: Dactylosporangium luridium sp. nov., Dactylosporangium luteum sp. nov. and Dactylosporangium salmoneum sp. nov. Int J Syst Evol Microbiol. doi:10.1099/ijs.0.016451-0
Koeppel A, Perry EB, Sckorski J, Krizane D, Warner A et al (2008) Identifying the fundamental units of bacterial diversity, a paradigm shift to incorporate ecology into bacterial systematics. Proc Natl Acad Sci USA 105:2504–2509
Konstantinidis KT, Tiedje JM (2005) Genomic insights into the species definition for prokaryotes. Proc Natl Acad Sci USA 102:2567–2572
Konstantinidis KT, Tiedje JM (2007) Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr Opin Microbiol 10:504–509
Kopke B, Wilins R, Engelen B, Cypionka H, Sass H (2005) Microbial diversity in coastal subsurface sediments: a cultivation approach using various electron acceptors and substrate gradients. Appl Environ Microbiol 71:7819–7830
Krieg NR (2005) Identification of prokaryotes. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds) Bergey’s manual of systematic bacteriology, 2nd edn, vol 2, part A, introductory essays. Springer, USA, pp 33–38
Kroppenstedt RM, Goodfellow M (2006) The family Thermomonosporaceae: Actinocorallia, Actinomadura, Spirillispora and Thermomonospora. In: Dworkin M, Falkow S, Schleifer KH, Stackebrandt E (eds) The prokaryotes, 3rd edn, vol 3, Archaea and Bacteria: Firmicutes, Actinomycetes. Springer, New York, pp 682–724
Kumar Y, Goodfellow M (2008) Five new members of the Streptomyces violaceusniger 16S rRNA gene clade: Streptomyces castelarensis comb. nov., S. humastatinus sp. nov., S. mordarskii sp. nov., S. rapamycinicus sp. nov. and S. ruanii sp. nov. Int J Syst Microbiol 58:1369–1378
Kumar Y, Goodfellow M (2010) Reclassification of Streptomyces hygroscopicus strains as Streptomyces aldersoniae sp. nov., Streptomyces augustmycinicus sp. nov., comb. nov. Streptomyces ascomycinicus sp. nov., Streptomyces decoyicus sp. nov., comb. nov., Streptomyces milbemycinicus sp. nov. and Streptomyces wellingtoniae sp. nov. Int J Syst Ecol Microbiol 60:769–775
Kumar Y, Aiemsun-ang P, Ward AC, Goodfellow M (2007) Diversity and geographical distribution of members of the Streptomyces violaceusniger 16S rRNA gene clade detected by clade specific primers. FEMS Microbiol Ecol 62:54–63
Kunisawa T (2007) Gene arrangements characteristic of the phylum Actinobacteria. Antonie van Leeuwenhoek 92:359–365
Kunst F, Ogasawara N, Mosyer I, Albertini AM, Alloni V et al (1997) The complete sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390:249–256
Kurahashi M, Tukanaga Y, Sakiyama Y, Harayama S, Yokota A (2009) Iamia majanohamensis gen. nov., sp. nov., an actinobacterium isolated from sea cucumber Holothuria edulis, and proposal of Iamiaceae fam nov. Int J Syst Evol Microbiol 59:869–873
Kurahashi M, Tukunaga Y, Sakiyama Y, Harayama S, Yokota A (2010) Euzebya tangerine gen. nov., a deeply branching actinobacterium isolated from the sea cucumber Halothuria edulis and proposal of Euzebyaceae fam. nov., Euzebyales ord. nov. and Nitriliruptoridae subclassis nov. Int J Syst Evol Microbiol. doi:10.1099/ijs.0.016543-0
Küster E, Williams ST (1964) Selective media for isolation of streptomycetes. Nature 202:928–929
Lam KS (2006) Discovery of novel metabolites from marine actinomycetes. Curr Opin Microbiol 9:245–251
Lapage SP, Sneath PHA, Lessel EF, Skerman VBD, Seeliger HPR, Clark WA (eds) (1975) International code of nomenclature of bacteria, 1975 revision. American Society for Microbiology, Washington, DC
Lapage SP, Sneath PHA, Lessel EF Jr, Skerman VBD, Seeliger HPR, Clark WA (eds) (1992) International code of nomenclature of bacteria (1990) revision: bacteriological code. American Society for Microbiology, Washington, DC
Larsen TO, Smedsgaard J, Nielson KF, Hansen ME, Frisvad JC (2005) Phenotypic taxonomy and metabolite profiling in microbial drug discovery. Nat Prod Rep 22:672–695
Lee SD (2008) Phycicococcus jejuensis gen. nov., sp. nov., an actinomycete isolated from seaweed. Int J Syst Evol Microbiol 56:2369–2373
Lee DW, Lee JM, Seo JP, Schumann P, Kim SJ, Lee SD (2008) Phycicola gilvas gen. nov., sp. nov., an actinobacterium isolated from living seaweed. Int J Syst Evol Microbiol 58:1318–1323
Li Hr, Yu Y, Luo W, Zeng Y-X (2010) Marisediminicola antarctica gen. nov., sp. nov., an actinobacterium isolated from the Antarctic. Int J Syst Evol Microbiol. doi:10.1099/ijs.0.018754-0
Liao Z-L, Tang S-K, Guo L, Zhang y-Q, Tian X-P, Jian C-L, Xu L-H, Li WJ (2009) Vewrrucosispora lutea sp. nov., isolated from a mangrove sediment sample. Int J Syst Evol Microbiol 59:2129–2379
Liu Z, Li Y, Zheng L-Q, Huang Y-J, Li W-J (2010) Saccharomonospora marina sp. nov., isolated from an ocean sediment in the East China Sea. Int J Syst Evol Microbiol. doi:10.1099/ijs.0.017038-0
Ludwig W, Klenk H-P (2005) Overview: a phylogenetic backbone and taxonomic framework for prokaryotic systematics. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds) Bergey’s manual of systematic bacteriology, 2nd edn, vol 2, the proteobacteria, part A, introductory essays. Springer, USA, pp 49–65
MacNaughton SJ, O’Donnell AG (1994) Tuberculostearic acid as a means of estimating the recovery (using dispersion and differential centrifugation) of actinomycetes from soil. J Microbiol Methods 20:69–77
Magarvey NA, Keller JM, Bernan V, Dworkin M, Skerman DH (2004) Isolation and characterization of novel marine-derived actinomycete taxa rich in bioactive metabolites. Appl Environ Microbiol 70:7520–7529
Maldonado LA, Stach JEM, Pathom-aree W, Ward AC, Bull AT, Goodfellow M (2005a) Diversity of cultivable actinobacteria in geographically widespread marine sediments. Antonie van Leeuwenhoek 87:11–18
Maldonado LA, Fenical W, Jensen PR, Kauffman CA, Mincer TJ, Ward AC, Bull AT, Goodfellow M (2005b) Salinispora arenicola gen. nov., sp. nov. and Salinispora tropica sp. nov., obligate marine actinomycetes belonging to the family Micromonosporaceae. Int J Syst Evol Microbiol 55:1759–1766
Maldonado LA, Stach JEM, Ward AC, Bull AT, Goodfellow M (2008) Characterisation of micromonosporae from aquatic environments using molecular taxonomic methods. Antonie van Leeuwenhoek 94:289–298
Maldonado LA, Frangoso-Yáñez D, Pérez-Garciä A, Rosellón-Druker J, Quintana E (2009) Actinobacterial diversity from marine sediments collected in Mexico. Antonie van Leeuwenhoek 95:111–120
Manfio GP, Atalan E, Zakrzewska-Czerwinska J, Mordarski M, Rodriguez C, Collins MD, Goodfellow M (2003) Classification of novel soil streptomycetes as Streptomyces aureus sp. nov., Streptomyces laceyi sp. nov. and Streptomyces sanglieri sp. nov. Antonie van Leeuwenhoek 83:245–255
McLeod MP, Warren H, Hsiao WW, Araki N, Myhre M et al (2006) The complete genome of Rhodococcus RHA 1 provides insights into a catabolic powerhouse. Proc Natl Acad Sci 103:15582–15587
McVeigh HP, Munro J, Embley TM (1996) Molecular evidence for the presence of novel actinomycete lineages in a temperate forest soil. J Ind Microbiol 17:197–204
Mehling A, Wehmeier UF, Piepersberg W (1995) Nucleotide sequences of streptomycete 16S ribosomal DNA—towards a specific identification system for streptomycetes using PCR. Microbiology 141:2139–2147
Mexson, J, Goodfellow M, Bull AT (2000) Preliminary studies of the diversity of micromonosporae in Indonesian soils and lake sediments. Int J Biotechnol Special Issue, June, pp 384–388
Mincer TJ, Jensen PR, Kaufmann CA, Fenical W (2002) Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl Environ Microbiol 68:5005–5011
Monciardini P, Sosio M, Cavaletti L, Chiochini C, Donadio S (2002) New PCR primers for the selective amplification of 16S rDNA from different groups of actinomycetes. FEMS Microbiol Ecol 42:419–429
Montalvo NF, Mohamed NM, Enticknap JJ, Hill RT (2005) Novel actinobacteria from marine sponges. Antonie van Leeuwenhoek 87:29–36
Morón R, Gonzalez I, Genilloud O (1999) New genus-specific primers for the PCR identification of members of the genera Pseudonocardia and Saccharoplyspora. Int J Syst Bacteriol 49:149–162
Newman DJ (2008) Natural products as leads to potential drugs: an old process or the hope for drug discovery? J Med Chem 61:2589–2599
Newman DJ, Cragg GM (2007) Natural products as sources of new drugs over the last 25 years. J Nat Prod 70:461–477
Oh D-C, Gontang EA, Kaufman CA, Jensen PR, Fenical W (2008) Salinipyrones and pacificanones, mixed-precursor polypeptides from the marine actinomycete Salinispora pacifica. J Nat Prod 71:570–575
Okoro CK, Brown R, Jones AL, Andrews BA, Asenjo JA, Goodfellow M, Bull AT (2009) Diversity of culturable actinomycetes in hyper-arid soils of the Atacama Desert, Chile. Antonie van Leeuwenhoek 95:121–133
Olano C, Méndez C, Salas JA (2009a) Antitumor compounds from marine actinomycetes. Mar Drugs 7:210–248
Olano C, Mèndez C, Salas JA (2009b) Antitumor compounds from actinomycetes from gene clusters to new derivatives by combinatorial synthesis. Nat Prod Rep 26:628–660
Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadley PJ (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL 23338. Nat Biotechnol 25:447–453
Ōmura S, Ikeda H, Ishikawa J et al (2001) Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci 98:12215–12220
Pathom-aree W, Nogi Y, Sutcliffe IC, Ward AC, Horikoshi K, Bull AT, Goodfellow M (2006a) Williamsia marianensis sp. nov., a novel actinomycete isolated from the Mariana Trench. Int J Syst Evol Microbiol 56:1123–1126
Pathom-aree W, Nogi Y, Sutcliffe IC, Ward AC, Horikoshi K, Bull AT, Goodfellow M (2006b) Dermacoccus abyssi sp. nov., a piezotolerant actinomycette isolated from the Mariana Trench. Int J Syst Evol Microbiol 56:1233–1237
Pathom-aree W, Nogi Y, Ward AC, Horikoshi K, Bull AT, Goodfellow M (2006c) Dermacoccus barathri sp. nov. and Dermacoccus profundi sp. nov., novel actinomycetes isolated from deep-sea mud of the Mariana Trench. Int J Syst Evol Microbiol 56:2303–2307
Pathom-aree W, Stach JEM, Ward AC, Horikoshi K, Bull AT, Goodfellow M (2006d) Diversity of actinomycetes isolated from Challenger Deep sediment (10, 898 m) from the Mariana Trench. Extremophiles 10:181–189
Payne DJ, Bradley J, Edwards JE, Gilbert D, Scheld, Bartlett JG (2007) Drugs from bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6:29–40
Penn K, Jenkins C, Nett M, Udwary DW, Gontang EA et al (2009) Genomic islands link secondary metabolism to functional adaptation in marine actinomycetes. ISMEJ 3:1193–1203
Piel J (2004) Metabolites from symbiotic bacteria. Nat Prod Rep 21:519–538
Pimentel-Elardo SM, Scheuermayer M, Kozytska S, Hentschel U (2009) Streptomyces axinellse sp. nov., isolated from the Mediterranean sponge Axinella polyporides (Poriferae). Int J Syst Evol Microbiol 59:1433–1437
Priest FG (2004) Approaches to identification. In: Bull AT (ed) Microbial diversity and bioprospecting. ASM Press, Washington, DC, pp 49–56
Priest FG, Goodfellow M (eds) (2000) Applied microbial systematics. Kluwer Academic Publishers, Dordrecht, pp 1–479
Qin S, Li J, Zhang Y-Q, Zhu W-Y, Zhao G-Z, Xu L-H, Li W-J (2009) Plantactinospora mayteni gen. nov., sp. nov., a member of the family Micromonosporaceae. Int J Syst Evol Microbiol 59:2527–2533
Qui D, Ruan J, Huang Y (2008) Selective isolation and identification of members of the genus Micromonospora. Appl Environ Microbiol 74:5593–5597
Riedlinger J, Reicke A, Zähner H, Krismer B, Bull AT, Maldonado LA, Ward AC, Goodfellow M, Bister B, Bischoff D, Süssmuth R, Fiedler H-P (2004) Abyssomicins, inhibitors of the para-aminobenzoic acid pathway produced by the marine Verrucosispora strain AB-18-032. J Antibiot 57:271–279
Rosselló-Mora R, Amann R (2001) The species concept for prokaryotes. FEMS Microbiol Rev 25:39–67
Rowbotham TJ, Cross T (1977) Ecology of Rhodococcus coprophilus and associated actinomycetes in freshwater and agricultural habitats. J Gen Microbiol 100:231–240
Salanoubat M, Genin F, Arliguenave F, Gouzy J, Mangenot S et al (2002) Genome sequence of the plant pathogen Ralstonia solanacearum. Nature 415:497–502
Salazar O, Morón R, Genilloud O (2000) New genus-specific primers for the PCR identification of members of the genus Saccharomonospora and evaluation of the diversity of wild-type isolates of Saccharomonospora detected from soil DNAs. Int J Syst Evol Microbiol 50:2043–2053
Salt M, Hugenholz P, Janssen PH (2002) Cultivation of globally distributed soil bacteria phylogenetic lineages previously only detected in cultivation-independent surveys. Environ Microbiol 4:654–656
Schleifer K-H (2010) Classification of Bacteria and Archaea: past, present and future. Syst Appl Microbiol 32:533–542
Schloss PD, Handelsman J (2005) Metagenomics for studying unculturable microorganisms: cutting the Gordian knot. Genome Biol 6:229–232
Schneider K, Keller S, Wolter FE, Röglin L, Beil W, Seitz O, Nicholson G, Bruntner C, Riedlinger J, Fiedler H-P, Süssmuth RD (2008) Proximicins A, B and C—antitumor furan analogues of netropsin from the marine actinomycete Verrucosispora induce upregulation of p53 and the cyclin kinase inhibitor p21. Angew Chem Int Ed 47:3258–3261
Schneider K, Nachtigall J, Hänchen A, Nicholson G, Goodfellow M, Süssmuth RD, Fiedler H-P (2009) Lipocarbazoles, new secondary metabolites from Tsukamurella pseudospumae Acta 1857 with antioxidative activity. J Nat Prod 72:1768–1772
Sembiring L, Ward AC, Goodfellow M (2000) Selective isolation and characterisation of members of the Streptomyces violaceusniger clade associated with the roots of Paraserianthes falcataria. Antonie van Leeuwenhoek 78:353–366
Seo YB, Kim D-E, Kim G-D, Kim H-W, Nam S-W, Kim YT, Lee HJ (2009) Kocuria givangalliensis sp. nov., an actinobacterium isolated from seawater. Int J Syst Evol Microbiol 59:2769–2772
Seviour RJ, Kragelund C, Long Y, Eales K, Nielsen JL, Nielsen PH (2008) Ecophysiology of actinobacteria in activated sludge systems. Antonie van Leeuwenhoek 94:21–33
Shen FT, Young C-C (2005) Rapid detection and identification of the metabolically diverse genus Gordonia by 16S rRNA-gene-targeted genus-specific primers. FEMS Microbiol Lett 250:221–227
Skerman VBD, McGowan V, Sneath PHA (1980) Approved lists of bacterial names. Int J Syst Bacteriol 30:225–420
Sneath PHA (2005) Bacterial nomenclature. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds) Bergey’s manual of systematic bacteriology, 2nd edn, vol 2, part A, introductory essays. Springer, USA, pp 83–88
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ (2006) Microbial diversity in the deep sea and the unexplored “rare biosphere”. Proc Natl Acad Sci USA 103:12115–12120
Stach JEM, Bull AT (2005) Estimating and comparing the diversity of marine actinobacteria. Antonie van Leeuwenhoek 87:3–9
Stach JEM, Maldonado LA, Ward AC, Goodfellow M, Bull AT (2003a) New primers specific for Actinobacteria: application to marine and terrestrial environments. Environ Microbiol 5:828–841
Stach JEM, Maldonado LA, Masson DG, Ward AC, Goodfellow M, Bull AT (2003b) Statistical approaches for estimating actinobacterial diversity in marine sediments. Appl Environ Microbiol 69:6189–6200
Stach JEM, Maldonado LA, Ward AC, Bull AT, Goodfellow M (2004) Williamsia maris sp. nov., a novel actinomycete isolated from the Sea of Japan. Int J Syst Evol Microbiol 54:191–194
Stackebrandt E, Schumann P (2006) Introduction to the taxonomy of Actinobacteria. In: Dworkin M, Falkow S, Schleifer RH, Stackebrandt RM (eds) The prokaryotes, 3rd edn, vol 3, Archaea and Bacteria: Firmicutes, Actinomycetes. Springer, New York, pp 297–321
Staley JT (2006) The bacterial species dilemma and the genomic-phylogenetic species concept. Philos Trans R Soc B 361:1899–1909
Stingl U, Cho J-C, Foo W, Vergin KL, Lanoil B, Giovannoni SJ (2008) Dilution-to-extinction of psychrotolerant planktonic bacteria from permanently ice-covered lakes in the McMurdo Dry Valleys, Antarctica. Microb Ecol 55:395–405
Stover CK, Pham XQ, Erwin AL, Mizoguishi SD, Warrener P et al (2000) Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406:959–964
Strohl WR (2004) Antimicrobials. In: Bull AT (ed) Microbial diversity and bioprospecting. ASM Press, Washington, DC, pp 336–355
Suzuki S-I, Okuda T, Komatsubara S (2001a) Selective isolation and distribution of the genus Planomonospora in soils. Can J Microbiol 47:253–263
Suzuki S-I, Okuda T, Komatsubara S (2001b) Selective isolation and study of the global distribution of the genus Planobispora in soils. Can J Microbiol 47:979–986
Talbot GH, Bradley J, Edwards JE Jr, Gilbert D, Scheld M, Bartlett JG (2006) Bad bugs need drugs: an update of the development pipeline from the antimicrobial task force of the Infectious Diseases Society of America. Clin Infect Dis 42:657–668
Tamura T, Ishida Y, Norzawa Y, Otoguro M, Suzuki K-I (2009) Transfer of Actinomadura spadix Nonomura and Ohara 1971 to Actinoallomurus spadix gen. nov., comb. nov., and description of Actinoallomurus amamiensis sp. nov., Actinoallomurus caesius sp. nov., Actinoallomurus coprocola sp. nov., Actinoallomurus fulvus sp. nov., Actinoallomurus iriomotensis sp. nov., Actinoallomurus luridus sp. nov., Actinoallomurus purpureus sp. nov. and Actinoallomurus yoronensis sp. nov. Int J Syst Evol Microbiol 59:1867–1874
Tan GYA, Robinson S, Lacey E, Goodfellow M (2006) Exploration of Amycolatopsis diversity in soil using genus-specific primers and novel selective media. Syst Appl Microbiol 29:557–569
Tan GYA, Robinson S, Lacey E, Brown W, Kim W, Goodfellow M (2007) Amycolatopsis regifaucium sp. nov., a novel actinomycete that produces kigamicins. Int J Syst Evol Microbiol 57:2562–2567
The Society for Actinomycetes Japan (2001) Identification manual of actinomycetes. Business Center for Academic Societies, Tokyo
Theobald U, Schimana J, Fiedler H-P (2000) Microbial growth and production kinetics of Streptomyces antibioticus Tu 6040. Antonie van Leeuwenhoek 78:307–313
Tian X-P, Tang S-K, Dong J-D, Zhang Y-Q, Xu L-H, Zhang S, Li W-J (2009a) Marinactinospora thermotolerans gen. nov., sp. nov., a marine actinomycete isolated from a sediment in the northern South China Sea. Int J Syst Evol Microbiol 59:948–952
Tian X-P, Zhi X-Y, Qiu Y-Q, Zhang Y-Q, Tang S-K, Xu L-H, Zhang S, Li W-J (2009b) Sciscionella marina gen. nov., sp. nov., a marine actinomycete isolated from a sediment in the northern South China Sea. Int J Syst Evol Microbiol 59:222–228
Tindall BJ, Roseselló-Mora R, Busse HJ, Ludwig W, Kämpfer P (2010) Notes on the characterization of prokaryotic strains for taxonomic purposes. Int J Syst Evol Microbiol 80:249–266
Trüper H (1999) How to name a prokaryote? Etymological considerations, proposals and practical advice in prokaryotic nomenclature. FEMS Microbiol Rev 23:231–249
Trüper H (2005) Etymology in nomenclature of prokaryotes. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds) Bergey’s manual of systematic bacteriology, 2nd edn, vol 2, the proteobacteria, part A, introductory essays. Springer, USA, pp 89–99
Udwary DW, Ziegler L, Asolbar HN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS (2007) Genome sequencing reveals complete secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci USA 104:10376–10381
Ue H, Matsuo Y, Kasai H, Yokota A (2010) Miniimonas arenae gen. nov., sp. nov., a novel actinobacterium isolated from sea sand. Int J Syst Evol Microbiol. doi:10.1099/ijs.0.019596-0
Vandamme P, Pot P, Gillis M, de Vos P, Kersters K, Swings J (1996) Polyphasic taxonomy: a consensus approach to bacterial systematics. Microbiol Rev 60:407–438
Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, van Sinderen D (2007) Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev 71:495–548
Vickers JC, Williams ST (1987) An assessment of plate inoculation procedures for the enumeration and selective isolation of streptomycetes. Microbios Lett 35:113–117
Vickers JC, Williams ST, Ross GW (1984) A taxonomic approach to selective isolation of streptomycetes from soil. In: Ortiz-Ortiz L, Bojalil LF, Yakoleff V (eds) Biological, biochemical and biomedical of actinomycetes. Academic Press, London, pp 553–561
Ward AC, Goodfellow M (2004) Phylogeny and functionality: taxonomy as a roadmap to genes. In: Bull AT (ed) Microbial diversity, bioprospecting. ASM Press, Washington, DC, pp 288–313
Watve MG, Tickoo R, Jog MM, Bhole BD (2001) How many antibiotics are produced by the genus Streptomyces. Arch Microbiol 176:386–390
Wayne LG, Brenner DJ, Colwell RR, Grimont PAD, Kandler O et al (1987) Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol 37:463–464
Whitman WB, Coleman DC, Wiebe WJ (1998) Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95:6578–6583
Williams PG (2008) Panning for chemical gold: marine bacteria as a source of new therapeutics. Trends Biotechnol 27:45–51
Williams ST, Vickers JC (1988) Detection of actinomycetes in the natural environment: problems and perspectives. In: Okami Y, Beppu T, Ogawara K (eds) Biology of actinomycetes. Japan Scientific Societies Press, Tokyo, pp 265–270
Williams ST, Davies FL, Hall DM (1969) A practical approach to the taxonomy of actinomycetes isolated from soil. In: Sheals JG (ed) The soil ecosystem. The Systematics Association, London, pp 107–117
Williams PG, Asolkar RN, Kondratyuk T, Pezzulo JM, Jensen PR, Fenical W (2007) Saliniketals A and B, bicyclic polyketides from the marine actinomycete Salinispora arenicola. J Nat Prod 70:83–88
Woese CR (1987) Bacterial evolution. Microbiol Rev 51:221–271
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E et al (2009) A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nat Lett 426:1056–1060
Xu J, Wang Y, Xie S-J, Xu J, Xiao J, Ruan J-S (2009) Streptomyces xiamenensis sp. nov., isolated from a mangrove sediment. Int J Syst Evol Microbiol 59:472–476
Yi H, Schumann P, Sohn K, Chun J (2004) Serinicoccus marinus gen. nov., sp. nov., a novel actinomycete with L-ornithine and L-serine in the peptidoglycan. Int J Syst Evol Microbiol 54:1585–1589
Yi H, Schumann P, Chun J et al (2007) Demequina aestuarii gen. nov., sp. nov., a novel actinomycete of the suborder Micrococcineae, and reclassification of Cellulomonas fermentaris Bagnara et al. 1985 as Actinotalea fermentaris gen. nov., comb. nov. Int J Syst Evol Microbiol 57:151–156
Yoon JH, Lee ST, Shin YK, Kim SB, Kim HJ, Goodfellow M, Park YH (1996) Rapid identification of Saccharomonospora strains by multiplex PCR using species-specific primers within the 16S rRNA gene. J Microbiol Methods 27:89–95
Zhang H, Lee YK, Zhang W, Lee HK (2006) Culturable actinobacteria from the marine sponge Hymeniacidon perleve: isolation and phylogenetic diversity by 16S rRNA gene-RFLP analysis. Antonie van Leeuwenhoek 90:159–169
Zhi X-Y, Li W-J, Stackebrandt E (2009) An update on the structure and 16S rRNA gene sequence-based definition of higher ranks of the class Actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the existing higher taxa. Int J Syst Evol Microbiol 59:589–608
Acknowledgments
We thank our co-workers and collaborators for their valuable contributions to papers cited in this article. We are also indebted to Alan Bull, Paul Jensen, Wen-Jun Li, Barny Whitman and Xiao-Yang Zhi for helpful comments on earlier versions of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Goodfellow, M., Fiedler, HP. A guide to successful bioprospecting: informed by actinobacterial systematics. Antonie van Leeuwenhoek 98, 119–142 (2010). https://doi.org/10.1007/s10482-010-9460-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-010-9460-2