
Abstract
Antiangiogenic therapy, specially sorafenib, has become the standard of care for patients with advanced hepatocellular carcinoma (HCC), however, the improvement in survival time is not satisfactory. Previous studies have found that, in some circumstances, antiangiogenic therapy promoted tumor metastasis and the mechanistic studies were mainly focus on cancer-cell-autonomous manners. In two experimental metastasis models with tail-vein injection with hepatoma cells and an orthotopic HCC mouse model, we found that pretreatment with two vascular endothelial growth factor receptor (VEGFR) inhibitors, sunitinib and sorafenib, facilitated tumor cell survival in blood stream and promoted lung metastasis from tumors that were subsequently incubated after drug discontinuation, indicating that host response joined into the pro-metastatic effects. An antibody microarray identified that interleukin (IL)-12b was decreased in the peripheral blood of the mice treated with the two VEGFR inhibitors. IL-12b suppression in macrophages and dendritic cells from host organs was found to play a crucial role in treatment-induced metastasis. Supplement with recombinant mouse IL-12b or restoration of IL-12b expression in the host by zoledronic acid, which was previously reported to enhance IL-12 expression in vitro and in vivo, alleviated the metastasis-promoting effects of sunitinib and sorafenib. These studies suggest that host response to VEGFR inhibitors facilitates HCC metastasis and restoration of IL-12b expression could translate into clinical benefits.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary liver cancer [hepatocellular carcinoma (HCC) in about 85 % of cases] is the second-greatest cause of cancer death in men and the sixth-greatest cause of cancer death in women worldwide [1]. Most patients with newly diagnosed HCC are not candidates for curative treatments [2]. Sorafenib, a tyrosine kinase inhibitor (TKI) with an antiangiogenic effect that mainly targets vascular endothelial growth factor receptors (VEGFRs), has become the standard of care for patients with advanced HCC. However, in 2 phase III clinical trials, SHARP [3] and Oriental study [4], sorafenib prolonged patients’ median survival time by <3 months, and most patients had to discontinue drug therapy for various reasons. Although the effect of discontinuation of sorafenib was not reported, rebound of tumor growth was reported in patients continuing or discontinuing treatment with other antiangiogenic drugs [5–7].
In clinical practice, the prolonged progression-free survival achieved with some antiangiogenic therapies does not always translate into an overall survival benefit [8]. Some “opposite” effects of antiangiogenic therapies, namely proinvasion and prometastasis effects, could counteract the antitumor effects. Clinical outcome would be influenced by both antitumor and prometastasis effects. Antiangiogenic therapies were found to facilitate tumor metastasis in preclinical studies [9, 10] showing an opposite effect. Studies on the mechanisms, including our recent research [11], were mainly focused on tumor cells response to treatment, often neglecting the internal host environment. More recently, effects of antiangiogenic therapy acted upon the host have been observed [12], and changes in the host or metastatic target organ sometimes facilitated metastasis [9, 13, 14]. However, the mechanism of host-mediated prometastasis effects and its potential of interventions remain largely unrevealed.
In order to treat the host without exposing tumor cells to antiangiogenic agents, we used the pretreatment schedule in this study. In the pretreatment schedule, drug administration was terminated before tumor incubation. In accordance with previous studies [9, 13, 14], we found that pretreatment with sunitinib and sorafenib, 2 VEGFR TKIs with a similar spectrum of kinase inhibition, facilitated lung metastasis of HCC cells. This prometastasis effect was primarily mediated by downregulation of interleukin (IL)-12b (also known as IL-12p40) and suppression of the cytotoxic effect of NK cells, suggesting that host immunosuppression is critical to the prometastasis effect of antiangiogenic therapy. We also found that zoledronic acid (ZA), which has been reported to enhance IL-12 expression in vitro and in vivo [15, 16], alleviated the prometastasis effects of these 2 antiangiogenic agents by restoring the downregulated IL-12b.
Materials and methods
Animals, cell lines and treatments
Male wide-type C57BL/6 mice and male athymic Balb/c nu/nu mice of 5-week old were obtained from Shanghai Institute of Materia Medica (Chinese Academy of Science, Shanghai, China). B6.129S1-Il12b tm1Jm /J mice, an IL-12b KO strain with the background strain of C57BL/6, were bought from Jackson Laboratories (Bar Harbor, Maine). All animal experimental protocols were approved by Shanghai Medical Experimental Animal Care Committee.
Three human hepatoma cell lines, HCCLM3, established by our institute [17]; HepG2 and PLC/PRF/5 (Chinese Academy of Sciences) were used to establish xenograft models. Stable red fluorescent protein (RFP)-transfected HCCLM3 (HCCLM3-RFP) cells and green fluorescent protein (GFP)-transfected HepG2 (HepG2-GFP) cells were established as described elsewhere [18]. Experimental metastatic models were established by tail-vein injection of human hepatoma cell suspension with 1 × 106 cells into nude mice or by injection of 5 × 105 Hepa1-6 (Shanghai Institute of Cell Biology), a murine hepatoma cell line, into IL-12b KO mice or wide-type C57BL/6 mice.
Mice were treated with sunitinib and sorafenib which were gifts from Bayer and Pfizer, respectively at the dosage of 100 mg/kg/day by gavage. Sunitinib and sorafenib were suspended in a vehicle (cremophor EL:ethanol:water = 12.5:12.5:75). For all the in vivo studies, this vehicle was administrated as the control group. ZA was purchased from Novartis and was administrated at the dosage of 100 μg/kg/day via i.p. injection. According to the user’s manual, 0.3–1 μg/mL monoclonal anti-mouse IL-12b antibody (R&D Systems, Inc., Minneapolis, MN) can neutralize 1 ng/mL recombinant mouse IL-12b (rmIL-12b). The plasma concentration of IL-12b was approximately 300 pg/mL, at most, for the nude mice (see “Results” for detail). Therefore, i.p. injection of IL-12b antibody was administered at the dosage of 6 μg per mouse, twice a week.
Evaluation of lung metastasis
Lungs from models established with HCCLM3-RFP or HepG2-GFP were excised, and a stereomicroscope imaging system (Leica Microsystems Imaging Solutions Ltd) was used to view metastatic foci with fluorescent protein. Metastatic foci exhibiting fluorescence were measured with Image-Pro Plus software (Media Cybernetics, Bethesda, MD) as described [19], and the surface area of the lungs was measured in bright field. Lung metastasis was expressed as the ratio of metastatic foci to the corresponding lung surface area.
Lungs from models established with cell lines without fluorescent protein transfection (PLC/PRF/5 and Hepa1-6) were excised, fixed with 4 % formaldehyde, and embedded in paraffin. Serial sections were cut into the thickness of 5 μm. Metastasis was identified on the lung sections with H&E staining. For each mouse, 30 randomly selected sections of lung were sampled, and the metastatic burden was assessed according to the size under the microscope (Leica CME, Leica Microsystems Imaging Solutions Ltd, Cambridge, UK). Metastatic foci with less than 5 cells, 6–15 cells, or more than 15 cells were allocated scores of 1, 2, or 3, respectively. The metastasis index was the sum of the scores. For metastatic foci occupying serial sections, only the largest cross-section was scored. The metastatic scores were evaluated independently by 2 investigators who were blinded to the groups.
Disseminating tumor foci in the lungs were evaluated on frozen sections to investigate the short-term fate of the tumor cells after incubation. Three days after tail-vein injection with HepG2-GFP cells, mice were sacrificed and frozen sections of the lungs were produced from 15 randomly selected sections. The slides were fully reviewed under bright field and GFP+ foci in all sections were counted under fluorescent field. An independent investigator blinded to group designations took photographs of the 3 fields (100×) with the most GFP+ foci (hotspots) for each mouse. The metastatic foci were then counted. The number of disseminating foci was counted as the number of total GFP+ foci per 100× field.
Antibody microarray and ELISA for the detection of plasma cytokines
Mice were anesthetized and the orbital blood was obtained. Plasma samples were collected and stored at −80 °C. The cytokines with differential expression were compared among different groups using a mouse cytokine antibody array 6 (RayBiotech, Norcross, GA) which included 97 specific cytokines and angiogenic factors, according to the manufacturer’s instructions. Plasma from 4 mice was pooled for antibody assay. The concentration of plasma IL-12b was detected by an ELISA assay (R&D Systems).
Separations of macrophages and dendritic cells (DCs)
Macrophages and DCs were isolated from the spleens of Balb/c mice with a magnetic-activated cell sorting (MACS) system with the magnetic beads coupled to mouse/human CD11b microbeads and mouse pan DC microbeads (Miltenyi Biotec, Auburn; CA), respectively, as previously described [20]. The spleens excised from 3 mice were pooled for isolation of macrophages or DCs.
Primers used in reverse transcriptase (RT)-PCR assay
The primers used for the amplification of genes were as follows: mouse il-12b (mil-12b), forward 5′-AGACATGGAGTCATAGGCTCTG-3′ and reverse 5′-CCATTTTCCTTCTTGTGGAGCA-3′; mouse il-23p19, forward 5′-ATGCTGGATTGCAGAGCAGTA-3′ and reverse 5′-ACGGGGCACATTATTTTTAGTCT-3′; mouse il-12p35, forward 5′-AAATGAAGCTCTGCATCCTGC-3′ and reverse 5′-TCACCCTGTTGATGGTCACG-3′; mouse GAPDH, forward 5′-GCACAGTCAAGGCCGAGAAT-3′ and reverse 5′-GCCTTCTCCATGGTGGTGAA-3′; human Il-12b, forward 5′-CCAAGAACTTGCAGCTGAAG-3′ and reverse 5′-TGGGTCTATTCCGTTGTGTC-3′; and human β-actin, forward 5′-CACCCTGAAGTACCCCATCG-3′ and reverse 5′-TGCCAGATTTTCTCCATGTCG-3′. Fold changes were expressed and compared in −ΔCt, and −ΔCt plus a natural number to gain a positive number.
In vitro cell proliferation and invasion assay
Cell proliferation was counted with a CCK-8 assay (Dojindo, Tokyo, Japan). Cell invasion assay was performed as described [19]. Hepatoma cells (HepG2 and HCCLM3) that migrated through the permeable membrane [Matrigel (BD Biosciences, San Jose, CA) and transwell chamber] in 72 h were counted under an inverted light microscope at 100× magnification.
Flow cytometry
The expression of IFN-γ by natural killer (NK) cells in the peripheral blood was determined by flow cytometer. The immunostaining protocol was modified from the cell surface immunofluorescence staining protocol and intracellular cytokine staining protocol (Biolegend, San Diego, CA). PE-labeled antimouse CD49b (BD Pharmingen, San Diego, CA) and APC-labeled antimouse IFN-γ (Biolegend) were used to determine NK cells and intracellular IFN-γ expression. Circulating tumor cells (CTCs) were also counted by flow cytometer, with GFP as a marker for mice that had received a tail-vein injection of HepG2-GFP cells. The GFP-positive cells were gated with identically processed blood from a mouse that had not been injected with tumor cells.
Statistical analysis
The average levels of continuous data were reported as mean ± SD and were compared by student’s t test unless otherwise specified, using SPSS for Windows v13.0 (SPSS, Inc.). Pearson χ2 test or Fisher’s exact test was used to compare qualitative data. P < 0.05 (two sided) was considered statically significant.
Results
Pretreatment with sunitinib or sorafenib accelerated tumor metastasis
Sunitinib and vehicle were administered for 1 week in Balb/c nu/nu nude mice (n = 6 for each group), then HCCLM3-RFP was orthotopically implanted into the murine livers. Seven weeks later, tumor growth was not affected by sunitinib pretreatment (Fig. 1a; tumor volume, 462.2 ± 163.4 mm3 in sunitinib group vs. 417.3 ± 116.4 mm3 in control; P = 0.521, Mann–Whitney U test), while lung metastasis was significantly increased (Fig. 1b; ratio of metastatic foci to lung surface area, 0.057 ± 0.074 vs. 0.006 ± 0.001; P = 0.015, Mann–Whitney U test).

Pretreatment with sunitinib or sorafenib promoted liver cancer lung metastasis. In an orthotopic human hepatoma model, sunitinib or vehicle (control) was administrated 1 week before the orthotopic implantation of red fluorescent protein-labeled HCCLM3 tissue (n = 6 for each group). After 7 weeks, tumor volume did not differ between sunitinib and control groups (a), whereas lung metastasis was more prominent in the mice pretreated with sunitinib (b). Red fluorescent protein-positive metastatic foci could be observed under fluorescence microscopes (upper panel; scale bar 2 mm); and summary data of metastatic foci showed statistically significant differences (lower panel). Note, the contrast and brightness were linearly adjusted to highlight the metastatic foci and the borders of the lungs were marked with white lines. c In an experimental metastasis model, pretreatment with sunitinib or sorafenib enhanced lung metastasis of tail-vein injected green fluorescent protein (GFP)-labeled HepG2 cells in nude mice. Representative photographs of lungs under bright field and fluorescence fields from each groups are shown (n = 8 for each group; upper panel) and quantification of lung metastasis showed that sunitinib or sorafenib treatment led to significantly more metastatic foci in the lungs (lower panel). d When Hepa1-6 hepatoma cells were injected into tail vein of wide-type or IL-12b KO C57BL/6 mice, pretreatment with sunitinib again promoted lung metastasis in wide-type mice but not in IL-12b KO mice. For the mice treated with vehicle (control), IL-12b KO mice had a high incidence of lung metastasis compared with wide-type mice. A short time (3 days) after tail-vein injection with GFP-labeled HepG2 cells, the GFP+ circulating tumor cells were counted by flow cytometer and expressed as the ratio of their counts to the counts of peripheral blood mononuclear cell (PBMC) (e) and GFP+ metastatic cell clusters (arrows) were counted on frozen sections of lung tissues (f). Representative dot plots of circulating tumor cell counting and lung sections under bright field and fluorescence fields (e and f, upper panels) from each group are shown (n = 6 for each group). Mice pretreated with sunitinib or sorafenib had more circulating tumor cells in peripheral blood and more metastatic foci in lungs in compare with the vehicle-treated control (e and f, lower panels). All error bars, SD. Compared with the control, *P < 0.05; # P > 0.05
Pretreatment with sunitinib or sorafenib also accelerated lung metastasis in experimental metastasis models. Balb/c nu/nu mice were pretreated with sunitinib, sorafenib, or vehicle (n = 8 for each group) for 1 week, followed by tail-vein injection of HepG2-GFP. Eight weeks later, metastasis was evaluated in the liver, abdomen, and lungs. Both sunitinib and sorafenib pretreatment significantly increased lung metastasis (Fig. 1c; ratio of metastatic foci to lung surface area, 0.133 ± 0.096 and 0.086 ± 0.074 vs. 0.005 ± 0.011; P = 0.009 and P = 0.025, respectively). The sunitinib and sorafenib groups were not significantly different with respect to lung metastasis (P = 0.365). Liver metastasis and peritoneum metastasis could not be observed in this model. We attempted to inject HCCLM3-RFP cells into the tail vein to establish another experimental metastasis model, but lung metastasis could rarely be observed, therefore lung metastasis was not evaluated.
To investigate whether this prometastasis effect is limited to nude mice, we treated immunocompetent C57BL/6 mice with sunitinib or vehicle. In these mice, sunitinib pretreatment also led to significantly higher rates of lung metastasis compared to pretreatment with vehicle (fourfold, 8/12 vs. 2/12, P = 0.013; Fig. 1d).
These findings, that pretreatment with antiangiogenic drugs enhanced tumor metastasis, are in agreement with previous reports [9, 13, 14]. In the pretreatment schedule, only the host organs were directly exposed to the antiangiogenic drugs; tumor tissue or tumor cells were excluded from direct exposure, in view of the short half-lives (<12 h in mice) of sunitinib and sorafenib in mice [21, 22]. We can conclude that drug treatment led to changes in the mice host, a non-cancer-cell-autonomous mechanism, and therefore promoted lung metastasis of the subsequently introduced hepatoma cells.
We next studied the short-term effect of sunitinib or sorafenib pretreatment on CTCs. Three days after tail-vein injection with HepG2-GFP cells, peripheral blood was collected for counting CTCs and lungs were cut into frozen sections for evaluation of disseminating cells. Pretreatment with sunitinib yielded more GFP+ CTCs in peripheral blood compared with the vehicle-treated control (n = 6 for each group; the ratio of GFP+ cells to total PBMC, 0.149 ± 0.073 % vs. 0.077 % ± 0.019, P = 0.042; Fig. 1e) and more metastatic foci in the lungs (n = 6 for each group; 3.5 ± 0.9 vs. 1.8 ± 0.8, P = 0.006; Fig. 1f). There were also more GFP+ CTCs and more metastatic foci in the lungs from the sorafenib group but the difference did not reach a statistical significance (P = 0.095 and P = 0.381, respectively; Fig. 1e, f). This finding indicated that pretreatment with sunitinib impaired host clearance of disseminating tumor cells in peripheral blood and lungs, and these effects were manifested shortly after incubation of the tumor cells.
Antiangiogenic therapy decreased host-derived IL-12b expression
To investigate the effect that antiangiogenic agents act upon the host, the antibody microarray was used to survey changes in a panel of cytokine and angiogenic factors in murine peripheral blood. The concentration of IL-12b, a subunit of IL-12 heterodimer, in the peripheral blood of Balb/c mice decreased compared with vehicle only (Fig. 2a). IL-12b was also quantitatively measured by ELISA in Balb/c nu/nu nude mice at 2 time points (day 1 and day 7) after drug discontinuation. Both sunitinib and sorafenib groups had significantly lower plasma concentration of IL-12b on day 1 (Fig. 2b; P = 0.002 and P = 0.008, respectively), whereas plasma IL-12b in the sunitinib and sorafenib group approached the same level as in the control on day 7 (Fig. 2b; P = 0.122 and P = 0.348, respectively).

Host-derived IL-12b was suppressed by antiangiogenic therapy. a Several cytokines in the plasma from tumor-free Balb/c mice (the plasma from 4 mice was pooled) were detected by an antibody microarray. Among these cytokines, plasma IL-12b was lowered by both sunitinib and sorafenib (solid line rectangle = IL-12b; dotted line rectangle = positive control) by 0.61-fold and 0.75-fold, respectively (lower panel). b IL-12b concentration was further quantified by ELISA and was found to be significantly lowered by sunitinib and sorafenib on day 1 after the drug discontinuation (n = 3 for each group). However, on day 7 after drug discontinuation, plasma IL-12b in the treated group rose to the level of control (n = 3 for each group). c The expression of il-12b was significantly downregulated by sunitinib in kidneys, spleen, and liver, but not in lungs and il-12b was most abundantly expressed in spleen (n = 4 for each group). d Splenectomy decreased the plasma IL-12b concentration compared to sham operation on day 3 after operation (“sham op,” control; n = 6 for each group). However, 35 days after splenectomy, plasma IL-12b rose to the level of the control (n = 4 for each group). e When dendritic cells (DC) and macrophages (MΦ) were isolated from the spleens of mice treated with sunitinib, sorafenib, or vehicle (control) (3 spleens from each group were pooled), il-12b expression in macrophages and DCs was found to be suppressed by sunitinib and sorafenib. In addition, il-12b expression in the cells remaining after separation of MΦ (“−MΦ”) and DCs (“−DC”), which was much lower than in the macrophages and DCs, was also suppressed. f Il-12b expression in THP-1–derived macrophages was also suppressed by sunitinib and sorafenib in a dose-dependent manner in vitro (unit, μmol/L). All error bars, SD; compared with the control, *P < 0.05; # P > 0.05
To identify the source of IL-12b, we studied the expression of il-12b in the parenchymatous organs by RT-PCR and found that the expression of il-12b was significantly reduced by sunitinib in the kidneys, spleen, and liver (P = 0.004, 0.005, and 0.006), but not in lungs (P = 0.278) (Fig. 2c). Expression of il-12b was highest in the spleen among these 4 organs, suggesting that spleen was the main source of plasma IL-12b. Results after splenectomy further supported this supposition. Plasma IL-12b concentrations were compared between the Balb/c nu/nu mice after splenectomy and those after sham operation (control). Three days after operation, IL-12b concentration in the mice underwent splenectomy was significantly lower than in control (Fig. 2d; n = 6 in each group; 221.5 ± 34.3 pg/mL vs. 285.2 ± 28.5 pg/mL; P = 0.006). However, 2 months after splenectomy, plasma IL-12b concentration reached the same level in control (n = 4 in each group; 273.7 ± 51.8 pg/mL vs. 245.6 ± 40.5 pg/mL; P = 0.425) mimicking the short term and long term IL-12b changes after the discontinuations of antiangiogenic agents (Fig. 2b).
Because phagocytes and DCs are the main producers of IL-12 [23], we isolated macrophages and DCs from murine spleens (3 spleens from each group were pooled) using a MACS assay to detect IL-12b expression in these cells. As shown in Fig. 2e, il-12b expression was much lower in the residual cells after isolation than that in macrophages or DCs, suggesting that macrophages and DCs were the main producers of IL-12b. Moreover, il-12b expression in all these cells was suppressed by sunitinib or sorafenib. In vitro studies also found that il-12b expression in THP-1–derived macrophages could be downregulated by sunitinib or sorafenib in a dose-dependent manner (Fig. 2f).
IL-12b suppression was responsible for prometastasis effects
We used several mouse models to determine whether host IL-12b suppression mediates the prometastasis effects of antiangiogenic agents. As aforementioned, splenectomy decreased plasma IL-12b concentration. First, we compared lung metastasis in mice that underwent splenectomy with those underwent sham operation and found that splenectomy yielded more lung metastasis foci from tail-vein injected PLC/PRF/5 human hepatoma cells than sham operation (n = 9 for each group, metastatic foci, 6.3 ± 5.3 vs. 2.1 ± 2.7; P = 0.039, Mann–Whitney U test; Fig. 3a). A specific neutralizing antibody against IL-12 was also used to explore the effects of IL-12b. A group of Balb/c nu/nu mice was treated with anti-mouse IL-12b neutralizing antibody or normal saline as control twice per week for 2 weeks, followed by tail-vein injection with PLC/PRF/5 cells. Four weeks later, mice treated with IL-12b antibody had a lower mean body weight (Fig. 3b; n = 10 for each group, 22.5 ± 3.7 g vs. 25.3 ± 1.2 g; P = 0.034) and more lung metastasis (Fig. 3b; metastatic foci = 6.6 ± 4.7 vs. 2.2 ± 2.6; P = 0.018) compared with control.

IL-12b suppression played a crucial role in the prometastasis effects mediated by antiangiogenic drugs. a Compared to sham operation (“sham op”), splenectomy promoted metastasis of tail-vein–injected PLC/PRF/5 hepatoma cells. Representative photographs of the lung sections with H&E staining from each group were shown in upper panels (scale bars 200 μm; n = 9 for each group) and quantification of the lung metastasis foci showed that splenectomy significantly promoted lung metastasis (lower panel). Mean body weight of mice treated with IL-12b–specific neutralizing antibody (IL-12 Ab) was significantly lower than that of the control (n = 10 for each group; b, upper panel) and metastasis of hepatoma cells were more prominent in the IL-12b Ab–treated mice (b, lower panel). c Combined sunitinib and recombinant mouse IL-12b (rmIL-12b; n = 8) reversed the prometastasis effects of sunitinib (n = 8) in experimental metastasis models (n = 10 for control group). d Three days after tail-vein injection with hepatoma cells, IFN-γ expression in NK cells was significantly reduced. Representative dot plots were shown (upper panels), and the quantitative analysis demonstrated that NK counts were not differ among the 3 groups (n = 6 for each group; lower left panel), whereas IFN-γ expression in NK cells were suppressed by sunitinib and sorafenib (lower right panel). All error bars, SD. Compared with the control, *P < 0.05; # P > 0.05
We also investigated whether a decrease in IL-12b enhances lung metastasis in immunocompetent mice. Lung metastasis after tail vein injection of Hepa1-6 cells was compared between IL-12b KO and wild-type C57BL/6 mice which were pretreated with or without sunitinib. IL-12b KO mice were found to have higher rates of lung metastasis compared with wide-type mice (9/15 vs. 2/12; P = 0.047, Fisher’s exact test) (Fig. 1d). In the IL-12b KO mice, pretreatment with sunitinib also induced higher rates of lung metastasis, but the difference was not statistically significant (9/10 vs. 9/15; P = 0.179, Fisher’s exact test) suggesting that the prometastasis effect of sunitinib was reduced in the IL-12b KO mice (1.5-fold) in compare with that in the wide-type C57BL/6 mice (fourfold, Fig. 1d).
We next investigated whether supplementary rmIL-12b (R&D Systems) could reverse the prometastasis effects of antiangiogenic therapy. We pretreated Balb/c nu/nu mice with sunitinib, vehicle, or combined sunitinib and rmIL-12b (i.p. injection, 200 ng/kg, twice per week) for a week (n = 8, 10, or 8, respectively for each group) followed by tail-vein injection with HepG2-GFP cells. As shown in Fig. 3c, rmIL-12b administration partly alleviated the prometastasis effects of sunitinib. Sunitinib pretreatment again promoted lung metastasis, while its combination with rmIL-12b decreased lung metastasis (ratio of metastatic foci to lung surface area: sunitinib, 0.226 ± 0.150 vs. sunitinib + rmIL-12b, 0.088 ± 0.114; P = 0.059). The difference between metastasis in the control and sunitinib + rmIL-12b groups was not statistically significant (P = 0.434; Fig. 3c).
Suppressing IL-12b probably promoted lung metastasis by inhibiting NK function
We then asked how suppression of IL-12b promoted lung metastasis. First, we tested whether rmIL-12b directly inhibited tumor cell proliferation and invasion in vitro. We incubated hepatoma cells or L02 (a human embryonic liver cell line) with various concentrations of rmIL-12b for 48 h. Recombinant IL-12b showed a mild anti-proliferative effect on hepatoma or liver cell lines (Figure S1), except the proliferation of HCCLM3 was significantly suppressed by high concentrations of rmIL-12b (≥500 pg/mL), which is about twofold greater than the concentration of IL-12b in murine peripheral blood. The invasion of HepG2 cells through Matrigel was barely affected by rmIL-12b (100 pg/mL). The rmIL-12b and control groups did not differ with respect to the number of cells that invaded through the Matrigel (cells per 100× field, 272.3 ± 29.2 vs. 245.5 ± 33.3; P = 0.271).
These results suggested that the antitumor effect of IL-12b may be mediated by an indirect mechanism rather than direct effects. The antitumor effects of IL-12 are reported to be mediated mainly by induced IFN-γ expression in NK cells, NKT cells, and T cells [24]. Because the prometastasis effects were verified in both athymic nude mice (T cell deficiency) and immunocompetent mice in the present study, we studied how NK cells responded to sunitinib or sorafenib treatment. The aforementioned murine blood collected to detect CTCs was used for flow cytometry. The counts of CD49+ NK cells were not significantly different among the 3 groups (Fig. 3d). However, sunitinib or sorafenib treatment downregulated IFN-γ expression in circulating NK cells (n = 3 for each group; this result was replicated in another cohort) by a factor of 0.45 and 0.51, respectively, although the differences were not statistically significant (Fig. 3d), suggesting that the cytotoxic effects of NK cells were impaired by sunitinib and sorafenib.
ZA compromised the prometastasis effects of antiangiogenic therapy
Zoledronic acid is a bisphosphonate with a macrophage-modulating effect and has been used to treat or prevent bone metastasis in cancer patients [25]. In a xenograft hepatoma model, we found that sorafenib treatment induced abundant infiltration of monocytes/macrophages in tumor tissue, while sorafenib and ZA had a synergistic antitumor effect [26]. In the present study, we tested ZA in combination with sunitinib or sorafenib, because ZA has been reported to enhance IL-12 expression in DCs [15] and to stimulate the production of IL-12 in the effusion fluid in the murine model of mesothelioma [16].
Balb/c nu/nu mice were randomized into 3 groups and treated for 1 week with sunitinib, sunitinib + ZA, or vehicle (n = 6 for each group). Mice were sacrificed and the expression of il-12b and its related subunits in the spleen were evaluated by RT-PCR. The results showed that ZA reversed the downregulation of il-12b in murine spleens (Fig. 4a). Downregulation of 2 related IL-12b subunits, namely, il-12p35 and il-23p19, was also partially reversed (Fig. 4a). In addition, although ZA treatment suppressed the proliferation of THP-1–derived macrophages at a high concentrations (30 μmol/L, Fig. 4b), ZA reversed the il-12b suppression by sunitinib in THP-1–derived macrophages in vitro. The reversal was most prominent at a low concentration (1 μmol/L; Fig. 4c).

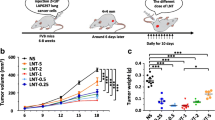
Zoledronic acid (ZA) reversed the expression of IL-12b and the prometastasis effects of antiangiogenic drugs. a ZA reversed the sunitinib-induced downregulation of il-12b in murine spleens. Downregulation of 2 related subunits of IL-12b, il-12p35 and il-23p19, was also partially reversed by zoledronic acid (compared with the control, *P < 0.05; # P > 0.05). b The proliferation of THP-1–derived macrophages was suppressed by ZA at high concentrations (30 μM), but was promoted at low concentrations (0.05 and 0.1 μM) (compared with 0 μmol/L, *P < 0.05; # P > 0.05). c The downregulation of il-12b by sunitinib in THP-1–derived macrophages was blunted by ZA at low concentration (0.2–5 μmol/L) (*P < 0.05 compared with control; # P < 0.05 compared with ZA at 0 μmol/l). ZA co-administration alleviated the prometastasis effects of sunitinib or sorafenib in an experimental metastatic model, in which, nude mice were injected with GFP-labeled HepG2 cells via tail vein (d, e). d Lungs from all the mice (n = 6 for each group) with metastatic foci were shown and e quantification of the lung metastasis foci demonstrated that ZA reversed the metastasis-promoting effects of sunitinib and sorafenib (compared with the control, *P < 0.05; # P > 0.05). All error bars, SD
Based on these findings, we treated Balb/c nu/nu nude mice with vehicle (control), monotherapy (sunitinib or sorafenib), or combination therapy (with ZA) for 1 week, followed by tail-vein injection of HepG2-GFP cells (n = 6 for each group). Seven weeks later, monotherapy with sunitinib or sorafenib again significantly enhanced lung metastasis (P = 0.028 and P = 0.036, respectively) which was in agreement with the data from Fig. 1c. Combination with ZA lowered the metastatic burden with a borderline statistical significance in compare with sunitinib or sorafenib monotherapy (P = 0.044 and P = 0.115, respectively), whereas the metastatic burdens of the combination therapy group and the control group were equal (P = 0.924 and P = 1.000, respectively; Fig. 4d, e), indicating that combination therapy with ZA can reverse the prometastasis effects of antiangiogenic therapy.
Discussion
In the present study, we found that sunitinib and sorafenib compromised the function of NK cells by downregulating host-derived IL-12b expression, resulting in an increased number of CTCs and lung metastases in murine models of HCC. ZA restored the expression of IL-12b and counterbalanced the prometastasis effect of sunitinib and sorafenib.
A number of studies have found that host response to systemic chemotherapy may potentially promote tumor metastasis, most of which was mediated by immunosuppression [27]. There are also reports that antiangiogenic treatment promoted invasiveness or metastasis by altering the tumor environment and/or tumor cells [11, 28, 29]. Ebos et al. [9] found that pretreatment with sunitinib or sorafenib accelerated tumor metastasis, suggesting that both agents induced changes in the microenvironment of mouse organs that are conditioned to be more permissive to cancer cell survival. Most recently, Chung et al. [14] found that sunitinib potentiated lung metastasis by increasing lung vascular permeability and cancer cell extravasation. On the other hand, preclinical and clinical studies have revealed that levels of cytokines and angiogenic factors in circulation were elevated by antiangiogenic drugs, including placental growth factor and VEGF, whereas levels of some cytokines with antiangiogenic features, e.g., soluble VEGFRs, decreased [30–32]. This indicates that the host environment became pro-angiogenic, probably compromising the efficacy of antiangiogenic therapy [12]. The present study added the evidence that the off-target effect of antiangiogenic drugs can modify the host immune system to be more permissive to circulating tumor cells. The clinical implication is that when antiangiogenic therapy is stopped, tumor growth will be unchecked by their influence and the prometastasis effects could become manifest. That could be one reason why many patients quickly relapse after discontinuing antiangiogenic treatment.
It has been reported that some VEGFR TKIs decreased IL-12 expression in human and in preclinical studies [32, 33]. Plasma IL-12 level was associated with resistance to antiangiogenic treatment [34]. Together, these findings imply that VEGFR TKIs may suppress host immunity by decreasing host IL-12b/IL-12 expression. It is not clear whether all VEGFR TKIs suppress host immunity, but evidence is continually emerging. There is a critical need for clinical studies because VEGFRs are among the most popular targets of molecular-targeted therapies.
Furthermore, the present study may indicate a way to overcome the opposite effect of VEGFR TKIs. As other authors have also reported [15, 16], we also proved that ZA treatment upregulated IL-12b expression. The chief clinical implication is that we should probably consider combining ZA, a proven anticancer drug, with VEGFR TKI therapies to counteract their potential prometastasis effects and improve the overall outcome. Based on results from the present and previous studies [26], we are conducting a phase II clinical trial (ClinicalTrials.gov identifier: NCT01259193) to test combination therapy.
This study has some potential limitations. Although the dosage of sunitinib or sorafenib (100 mg/kg/day) has been widely used in preclinical studies [35, 36], this dosage is relatively higher than that used in human patients, and the treatment-induced host changes were dose-dependent [13, 36]. Whether HCC patients who receive sorafenib treatment have similar changes remains to be seen. Second, because of the limitations of antibody array we used in this study, we cannot rule out the possibility that other cytokines which were not covered in the antibody array may also contribute to the increased rate of metastasis.
In conclusion, host responses to antiangiogenic therapy warrant more investigation in clinical settings and this response may be not unique to HCC. Counteracting this opposite effect could improve the clinical efficacy of antiangiogenic treatment.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61(2):69–90. doi:10.3322/caac.20107
Poon D, Anderson BO, Chen LT, Tanaka K, Lau WY, Van Cutsem E, Singh H, Chow WC, Ooi LL, Chow P, Khin MW, Koo WH (2009) Management of hepatocellular carcinoma in Asia: consensus statement from the Asian Oncology Summit. Lancet Oncol 10(11):1111–1118. doi:10.1016/S1470-2045(09)70241-4
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359(4):378–390. doi:10.1056/NEJMoa0708857
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J, Wang J, Tak WY, Pan H, Burock K, Zou J, Voliotis D, Guan Z (2009) Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 10(1):25–34. doi:10.1016/S1470-2045(08)70285-7
Cacheux W, Boisserie T, Staudacher L, Vignaux O, Dousset B, Soubrane O, Terris B, Mateus C, Chaussade S, Goldwasser F (2008) Reversible tumor growth acceleration following bevacizumab interruption in metastatic colorectal cancer patients scheduled for surgery. Ann Oncol 19(9):1659–1661. doi:10.1093/annonc/mdn540
Allegra CJ, Yothers G, O’Connell MJ, Sharif S, Petrelli NJ, Colangelo LH, Atkins JN, Seay TE, Fehrenbacher L, Goldberg RM, O’Reilly S, Chu L, Azar CA, Lopa S, Wolmark N (2011) Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C-08. J Clin Oncol 29(1):11–16. doi:10.1200/JCO.2010.30.0855
Powles T, Blank C, Chowdhury S, Horenblas S, Peters J, Shamash J, Sarwar N, Boleti E, Sahdev A, O’Brien T, Berney D, Beltran L, Nathan P, Haanen J, Bex A (2011) The outcome of patients treated with sunitinib prior to planned nephrectomy in metastatic clear cell renal cancer. Eur Urol 60(3):448–454. doi:10.1016/j.eururo.2011.05.028
Ebos JM, Kerbel RS (2011) Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 8(4):210–221. doi:10.1038/nrclinonc.2011.21
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS (2009) Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15(3):232–239. doi:10.1016/j.ccr.2009.01.021
Loges S, Mazzone M, Hohensinner P, Carmeliet P (2009) Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell 15(3):167–170. doi:10.1016/j.ccr.2009.02.007
Zhang W, Sun HC, Wang WQ, Zhang QB, Zhuang PY, Xiong YQ, Zhu XD, Xu HX, Kong LQ, Wu WZ, Wang L, Song TQ, Li Q, Tang ZY (2012) Sorafenib downregulates expression of HTATIP2 to promote invasiveness and metastasis of orthotopic hepatocellular carcinoma tumors in mice. Gastroenterology. doi:10.1053/j.gastro.2012.08.032
Kerbel RS, Ebos JM (2010) Peering into the aftermath: the inhospitable host? Nat Med 16(10):1084–1085. doi:10.1038/nm1010-1084
Welti JC, Powles T, Foo S, Gourlaouen M, Preece N, Foster J, Frentzas S, Bird D, Sharpe K, van Weverwijk A, Robertson D, Soffe J, Erler JT, Pili R, Springer CJ, Mather SJ, Reynolds AR (2012) Contrasting effects of sunitinib within in vivo models of metastasis. Angiogenesis. doi:10.1007/s10456-012-9291-z
Chung AS, Kowanetz M, Wu X, Zhuang G, Ngu H, Finkle D, Komuves L, Peale F, Ferrara N (2012) Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J Pathol 227(4):404–416. doi:10.1002/path.4052
Fiore F, Castella B, Nuschak B, Bertieri R, Mariani S, Bruno B, Pantaleoni F, Foglietta M, Boccadoro M, Massaia M (2007) Enhanced ability of dendritic cells to stimulate innate and adaptive immunity on short-term incubation with zoledronic acid. Blood 110(3):921–927. doi:10.1182/blood-2006-09-044321
Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Hegmans JP, Aerts JG (2010) Zoledronic acid impairs myeloid differentiation to tumour-associated macrophages in mesothelioma. Br J Cancer 103(5):629–641. doi:10.1038/sj.bjc.6605814
Li Y, Tang Y, Ye L, Liu B, Liu K, Chen J, Xue Q (2003) Establishment of a hepatocellular carcinoma cell line with unique metastatic characteristics through in vivo selection and screening for metastasis-related genes through cDNA microarray. J Cancer Res Clin Oncol 129(1):43–51. doi:10.1007/s00432-002-0396-4
Yang BW, Liang Y, Xia JL, Sun HC, Wang L, Zhang JB, Tang ZY, Liu KD, Chen J, Xue Q, Gao DM, Wu WZ (2008) Biological characteristics of fluorescent protein-expressing human hepatocellular carcinoma xenograft model in nude mice. Eur J Gastroenterol Hepatol 20(11):1077–1084. doi:10.1097/MEG.0b013e3283050a67
Zhu XD, Zhang JB, Fan PL, Xiong YQ, Zhuang PY, Zhang W, Xu HX, Gao DM, Kong LQ, Wang L, Wu WZ, Tang ZY, Ding H, Sun HC (2011) Antiangiogenic effects of pazopanib in xenograft hepatocellular carcinoma models: evaluation by quantitative contrast-enhanced ultrasonography. BMC Cancer 11(1):28. doi:10.1186/1471-2407-11-28
Xiong YQ, Sun HC, Zhang W, Zhu XD, Zhuang PY, Zhang JB, Wang L, Wu WZ, Qin LX, Tang ZY (2009) Human hepatocellular carcinoma tumor-derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin Cancer Res 15(15):4838–4846. doi:10.1158/1078-0432.ccr-08-2780
Haznedar JO, Patyna S, Bello CL, Peng GW, Speed W, Yu X, Zhang Q, Sukbuntherng J, Sweeny DJ, Antonian L, Wu EY (2009) Single- and multiple-dose disposition kinetics of sunitinib malate, a multitargeted receptor tyrosine kinase inhibitor: comparative plasma kinetics in non-clinical species. Cancer Chemother Pharmacol 64(4):691–706. doi:10.1007/s00280-008-0917-1
Hipp MM, Hilf N, Walter S, Werth D, Brauer KM, Radsak MP, Weinschenk T, Singh-Jasuja H, Brossart P (2008) Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood 111(12):5610–5620. doi:10.1182/blood-2007-02-075945
Trinchieri G (2003) Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 3(2):133–146. doi:10.1038/nri1001
Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, Anichini A (2007) Interleukin-12: biological properties and clinical application. Clin Cancer Res 13(16):4677–4685. doi:10.1158/1078-0432.CCR-07-0776
Lipton A (2011) Zoledronic acid: multiplicity of use across the cancer continuum. Expert Rev Anticancer Ther 11(7):999–1012. doi:10.1586/era.11.71
Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, Kong LQ, Wang L, Wu WZ, Tang ZY (2010) Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res 16(13):3420–3430. doi:10.1158/1078-0432.ccr-09-2904
Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, Norton L, Brogi E, Massague J (2012) A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150(1):165–178. doi:10.1016/j.cell.2012.04.042
Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, Isachenko N, Fouse SD, Phillips JJ, Cheresh DA, Park M, Bergers G (2012) VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22(1):21–35. doi:10.1016/j.ccr.2012.05.037
De Bock K, Mazzone M, Carmeliet P (2011) Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev Clin Oncol 8(7):393–404. doi:10.1038/nrclinonc.2011.83
Bagley RG, Ren Y, Weber W, Yao M, Kurtzberg L, Pinckney J, Bangari D, Nguyen C, Brondyk W, Kaplan J, Teicher BA (2011) Placental growth factor upregulation is a host response to antiangiogenic therapy. Clin Cancer Res 17(5):976–988. doi:10.1158/1078-0432.CCR-10-2687
Zhu AX, Sahani DV, Duda DG, di Tomaso E, Ancukiewicz M, Catalano OA, Sindhwani V, Blaszkowsky LS, Yoon SS, Lahdenranta J, Bhargava P, Meyerhardt J, Clark JW, Kwak EL, Hezel AF, Miksad R, Abrams TA, Enzinger PC, Fuchs CS, Ryan DP, Jain RK (2009) Efficacy, safety, and potential biomarkers of sunitinib monotherapy in advanced hepatocellular carcinoma: a phase II study. J Clin Oncol 27(18):3027–3035. doi:10.1200/JCO.2008.20.9908
Hanrahan EO, Lin HY, Kim ES, Yan S, Du DZ, McKee KS, Tran HT, Lee JJ, Ryan AJ, Langmuir P, Johnson BE, Heymach JV (2010) Distinct patterns of cytokine and angiogenic factor modulation and markers of benefit for vandetanib and/or chemotherapy in patients with non-small-cell lung cancer. J Clin Oncol 28(2):193–201. doi:10.1200/JCO.2009.22.4279
Yamamoto M, Kikuchi H, Ohta M, Kawabata T, Hiramatsu Y, Kondo K, Baba M, Kamiya K, Tanaka T, Kitagawa M, Konno H (2008) TSU68 prevents liver metastasis of colon cancer xenografts by modulating the premetastatic niche. Cancer Res 68(23):9754–9762. doi:10.1158/0008-5472.CAN-08-1748
Bhatt RS, Wang X, Zhang L, Collins MP, Signoretti S, Alsop DC, Goldberg SN, Atkins MB, Mier JW (2010) Renal cancer resistance to antiangiogenic therapy is delayed by restoration of angiostatic signaling. Mol Cancer Ther 9(10):2793–2802. doi:10.1158/1535-7163.mct-10-0477
Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C (2006) Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 66(24):11851–11858. doi:10.1158/0008-5472.CAN-06-1377
Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS (2007) Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci USA 104(43):17069–17074. doi:10.1073/pnas.0708148104
Acknowledgments
This work was jointly supported by Shanghai Natural Science Foundation (12ZR1442300), National Natural Science Foundation of China (No. 81020108025 and 81101564), the National Key Project for Infectious Diseases (2012ZX10002-012), and the National “211” Project for Higher Education.
Author information
Authors and Affiliations
Corresponding author
Additional information
Xiao-Dong Zhu and Hui-Chuan Sun have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhu, XD., Sun, HC., Xu, HX. et al. Antiangiogenic therapy promoted metastasis of hepatocellular carcinoma by suppressing host-derived interleukin-12b in mouse models. Angiogenesis 16, 809–820 (2013). https://doi.org/10.1007/s10456-013-9357-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-013-9357-6