
Abstract
Purpose
The purpose of these extensive non-clinical studies was to assess pharmacokinetics and dispositional properties of sunitinib and its primary active metabolite (SU12662).
Methods
Sunitinib was administered in single and repeat oral doses in mice, rats, and monkeys. Assessments were made using liquid-chromatography–tandem mass spectrometric methods, radioactive assays, and quantitative whole body autoradiography.
Results
Sunitinib was readily absorbed with good oral bioavailability and linear kinetics at clinically-relevant doses. SU12662 plasma levels were less than those of sunitinib in mice and monkeys, but greater in rats. Sunitinib was extensively distributed with moderate-to-high systemic clearance and eliminated primarily into feces. Single- and repeat-dosing kinetics were similar. A prolonged half-life allowed once-daily dosing, enabling adequate systemic exposure with limited-to-moderate accumulation. In multiple-dose studies with cyclic dosing, drug plasma concentrations cleared from one cycle to the next.
Conclusions
Sunitinib exhibited advantageous pharmacokinetic and dispositional properties in non-clinical species, translating into favorable properties in humans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sunitinib malate (SUTENT®; Pfizer Inc., New York, NY, USA) is an oral multitargeted tyrosine kinase inhibitor that selectively inhibits vascular endothelial growth factor receptors (VEGFR-1, -2, and -3), platelet-derived growth factor receptors (PDGFR-α and -β), stem cell factor receptor (KIT), Fms-like tyrosine kinase-3 receptor (FLT3), and the glial cell-line derived neurotrophic factor receptor (REarranged during Transfection, RET) ([1–4], Pfizer Inc., data on file). The United States (US) Food and Drug Administration and the European Commission have granted approval for the use of sunitinib in advanced renal cell carcinoma (RCC) and gastrointestinal stromal tumor (GIST) after disease progression on, or intolerance to, imatinib mesylate therapy. In addition, the compound is in development for the treatment of patients with other solid tumors, including neuroendocrine, breast, lung, colorectal, liver and prostate tumors [5–10].
Pharmacokinetic evaluations in humans have shown that maximum plasma concentrations of sunitinib are generally observed 6–12 h after oral administration, and that systemic exposure increases dose-proportionally over the dose range 25–100 mg [11]. Binding to human plasma proteins in vitro is 95% for sunitinib (and 90% for its primary active metabolite, SU12662), and independent of concentration (range studied: 100–4,000 ng/mL); the apparent volume of distribution for sunitinib is 2,230 L [11]. Sunitinib is metabolized primarily by cytochrome P450 (CYP3A4) to form SU12662, which comprises 23–37% of the total exposure and is further metabolized by CYP3A4 [12, 13]. Elimination of sunitinib is mainly via feces (61% of the administered dose [14]). Total oral clearance ranges from 34 to 62 L/h, and the terminal half-lives of sunitinib and SU12662 ranged from 40 to 60 h and 80 to 110 h, respectively, after a single oral dose [11]. Following repeat-dose administration, sunitinib accumulates 3- to 4-fold, while SU12662 accumulates 7- to 10-fold; steady-state concentrations are achieved within 10–14 days [11]. Food does not affect the disposition of sunitinib [15]. The pharmacokinetics are not affected in patients with Child-Pugh Class A and B hepatic impairment nor in patients with renal impairment, not on dialysis [11, 16].
Mendel et al. [2] reported a time- and dose-dependent pharmacokinetic/pharmacodynamic relationship between sunitinib exposure and antitumor activity. In several mouse xenograph models (representing tumors such as glioma and melanoma, and colon, epidermoid, non-small cell lung, and breast cancer), sunitinib doses of 20–80 mg/kg led to tumor regression or stasis. Specifically, Western blot analysis in athymic mice bearing A375 melanoma tumors [which express FLK1/KDR (VEGFR-2)] or SF767T glioma tumors (which express PDGFR-β) revealed that sunitinib 40 mg/kg inhibited the receptor phosphorylation of VEGFR-2 and PDGFR-β. Serial pharmacokinetic measurements indicated that bound and unbound sunitinib plasma concentrations at the 40 mg/kg dose were >50, which are the concentrations predicted from chemical- and cellular-based assays to inhibit VEGFR-2 and PDGFR-β. These experiments were the first to demonstrate a sunitinib exposure–response relationship, and led to the characterization of sunitinib pharmacokinetics and further investigation of the pharmacokinetic/pharmacodynamic relationship.
To date, only limited details of non-clinical pharmacokinetics across species for sunitinib and SU12662 have been published [17]. Here, we provide a comprehensive account of the pharmacokinetics and dispositional properties of both compounds based on the results of several studies in non-clinical species, including mice, rats, and Cynomolgus monkeys. These studies were part of the non-clinical development program that facilitated and supported the planning and implementation of clinical studies with sunitinib.
Materials and methods
Chemicals
Sunitinib (free base) and sunitinib l-malate (radiolabeled and non-radiolabeled) were obtained from SUGEN, Pharmacia, and/or Pfizer Inc. The free base was administered to animals in early studies, and sunitinib l-malate (the active ingredient in SUTENT®) was used in all pivotal studies. The l-malate salt is hereafter referred to as sunitinib, since bioequivalence was demonstrated for these different formulations (Pfizer Inc., data on file). All doses and concentrations are expressed as the free base equivalent of the drug.
General-purpose reagents and solvents were of analytical grade (or a suitable alternative), and were obtained principally from Sigma Chemical Company (St Louis, MO, USA), Fisher Scientific Company (Fair Lawn, NJ, USA), Carlo Erba Reagents, Sigma-Aldrich S.r.l. (Milan, Italy), and other approved vendors.
Animals and procedures
A number of single-dose and repeat-dose studies were conducted in mice, rats, and Cynomolgus monkeys. Monkeys were chosen for non-clinical assessment of safety and pharmacokinetics based on similarities in sunitinib metabolism to human beings (Pfizer Inc., data on file). The detailed procedures for typical in vivo studies in each of these species are described below. Pivotal studies were conducted in accordance with the generally recognized principles of Good Laboratory Practice, and procedures for all studies were in full compliance with the Animal Welfare Act and Regulations.
Female athymic mice, and male and female Sprague-Dawley rats were obtained from Charles River Laboratories (Calco-Lecco, Italy and Hollister, CA, Raleigh, NC, or Portage, MI, USA). Male and female Cynomolgus monkeys were obtained from Charles River Laboratories (Houston, TX, USA or CRPL, Mauritius). The test animals were fed with standard diets, except that food was withdrawn when the study protocols required fasting before dose administration. Water was available ad libitum in all studies.
Studies in mice
Single-dose administration. A single dose of sunitinib was administered intravenously or orally. For the intravenous route, sunitinib was prepared in an aqueous formulation (0.2 and 2.0 mg/mL in a vehicle containing: 1% hydrochloric acid, NF; 0.08% citric acid monohydrate, USP; 0.04% trisodium citrate dihydrate, USP; 0.5% polysorbate 80, NF; 10% polyethylene glycol 300, NF; and adjusted to pH 3.5 with sodium hydroxide, NF) for administration via the tail vein at doses of 1 or 10 mg/kg; blood samples were collected via cardiac puncture at 1, 5, 15, and 30 min, and 1, 3, 6, 12, 18, and 24 h post-dose. For the oral route, sunitinib was prepared in a carboxymethylcellulose (CMC) formulation (4, 8, 16, and 32 mg/mL in an aqueous vehicle containing: 0.5% CMC, NF; 0.9% sodium chloride, NF; 0.4% polysorbate 80, NF; and 0.9% benzyl alcohol, NF) and administered via gavage at doses of 20, 40, 80, or 160 mg/kg; blood samples were collected via cardiac puncture at 5, 15, and 30 min, and 1, 3, 6, 12, 18, and 24 h post-dose. Blood samples were maintained on ice until centrifugation to harvest plasma. After separation, plasma samples were stored at −80°C until analysis. All samples were protected from light.
Repeat-dose administration. Sunitinib, at therapeutic [40 mg/(kg day)] and supratherapeutic [80 mg/(kg day)] doses, was administered by oral gavage for 5 weeks to female SKH1 mice. Blood samples were collected 3 h after dosing on days 7, 9, 13, 20, and 34 for evaluation of systemic exposure. Samples were centrifuged and harvested plasma was stored at −20°C and protected from light until analysis.
Studies in rats
Single-dose administration. A single dose of sunitinib was administered intravenously or orally. For the intravenous route, sunitinib in an aqueous formulation (0.25, 1, and 2 mg/mL in the same vehicle used in studies in mice) was administered as a short 1-min infusion via a jugular vein catheter at doses of 1, 4, or 8 mg/kg; blood samples were collected via the carotid artery catheter at 2, 15, and 30 min, and 1, 3, 6, 9, and 24 h post-dose. For the oral route, sunitinib was prepared in a CMC formulation (0.5, 2.5, and 5 mg/mL in the same vehicle used in studies in mice) and administered via oral gavage at doses of 2, 10, or 20 mg/kg; blood samples were collected via the carotid artery at 5, 15, and 30 min, and 1, 3, 6, 9, and 24 h post-dose. Blood samples were maintained on ice until centrifugation to obtain plasma. All samples were protected from light and stored at −80°C until analysis.
Several single-dose studies in rats involved the administration of radiolabeled sunitinib formulations. In a typical study, solutions of [14C]-sunitinib were administered intravenously or orally. For the intravenous route, a dose of 5 mg/kg (4 mg/mL in water for injection, 1.48 MBq/mL) was given by bolus injection into the tail vein. An oral dose of 15 mg/kg (4 mg/mL in water for injection, 493 kBq/mL) was given by gastric gavage. Blood samples were collected via a superior vena cava cannula at the following time points: pre-dose, 5 and 20 min, and 1, 3, 6, 24, 48, and 72 h post intravenous dose; pre-dose, and 1, 3, 6, 8, 24, 48, and 72 h post oral dose. For mass balance studies, the rats were kept in metabolic cages, and urine and feces were also collected before dose administration and up to 72 h post-dose. An aliquot of blood was transferred to a paper cone for combustion and determination of 14CO2 radioactivity. The remaining blood was centrifuged to harvest plasma. An aliquot of plasma was transferred to a scintillation vial for measurement of drug-related radioactivity. The remaining plasma samples, together with urine and feces, were kept at −80°C until further analysis. All samples were protected from light.
Repeat-dose administration. In two non-clinical safety studies, sunitinib was administered orally for 3 or 6 months. In the 3-month study, male and female rats were treated with sunitinib 1.5, 5, or 15 mg/(kg day) for 91 days. In the 6-month study, sunitinib 0.3, 1.5, or 6 mg/(kg day) was administered for 4 weeks followed by no drug treatment for 1 week in each of five cycles (4/1 schedule). For these repeat-dose studies (and a third study in which rats were treated for 28 days), sunitinib was prepared in water for injection and administered via gavage.
Blood samples were typically collected pre-dose, and 1, 3, 6, 9, and 24 h post-dose on the first and last day of dosing, with collection of additional samples on days in between in some studies. For example, samples were collected on days 1, 28, 65 (early termination of drug treatment was necessary for the high-dose animals), and 91 of the 3-month study. For the 6-month study, full pharmacokinetic profiles were evaluated on days 1 and 28 (first treatment cycle) and on day 168 (the last day of the fifth treatment cycle); single-time point pharmacokinetic samples (pre-dose or 24 h post-dose) were also collected on days 36, 63, 71, 98, 106, 133, and 141, as well as on days 175, 196, and 224 during an 8-week recovery period. Harvested plasma was stored at –80°C and protected from light until analysis.
Studies in monkeys
Single-dose administration. In an initial study, two female Cynomolgus monkeys were dosed orally (nasogastric intubation; 50 mg/kg) and intravenously (5-min infusion into femoral vein; 2 mg/kg) with sunitinib in an aqueous vehicle [containing 0.5% (w/v) CMC sodium, 0.9% USP; sodium chloride, USP; 0.4% Tween 80, NF; and 0.9% benzyl alcohol, NF]. Blood samples were collected pre-dose and 0.5, 1, 1.5, 2, 3, 6, 9, and 24 h post oral dose administration from a saphenous vein. For intravenous studies, additional samples were collected at the end of the 5-min infusion and at 0.25 h post intravenous dosing. In a second study, sunitinib was prepared in an aqueous formulation [PEG200:saline, 5:95 (v/v)] for administration via bolus injection to male monkeys at a dose of 3 mg/kg. Blood samples were collected at various time points from pre-dose to 984 h post-dose. Single-dose pharmacokinetics of sunitinib and SU12662 were also evaluated on day 1 of a 28-day study in male monkeys, in which sunitinib in water was administered intravenously [3 mg/(kg day) via a saphenous vein] or orally [6 mg/(kg day) via nasogastric intubation] once daily—or once every other day (oral dose only)—for 28 days. Blood samples were collected from a femoral vein from pre-dose to 24 h post-dose. Blood samples were maintained on ice prior to centrifugation to obtain plasma. All samples were stored frozen at −70°C and protected from light prior to analysis.
In a study involving the oral administration of radiolabeled sunitinib, [14C]-sunitinib in solution in water for injection was administered at 6 mg/kg (4 mg/mL, 924 kBq/mL) by gastric gavage. Blood, urine, and feces were collected before dose administration and up to 336 h post-dose. These samples were processed and stored using the same procedures as in the [14C]-sunitinib study in rats described above.
Repeat-dose administration. A number of repeat-dose studies were conducted with sunitinib in Cynomolgus monkeys. These included once-daily dosing for 3 months [2, 6, and 20/12 mg/(kg day)] and 9 months [0.3, 1.5, and 6 mg/(kg day); five cycles of 4/1 schedule]. In the 3-month study, the highest dose group started with a dose of 20 mg/(kg day), but this was reduced to 12 mg/(kg day) from day 29 to the end of the study. For these studies, sunitinib was prepared in sterile water and administered via gavage.
Blood samples were typically collected over 24 h on the first and last days of dosing during the study period or 4-week dosing cycle. In some studies, samples were collected on additional days. For example, in the 3-month study, samples were collected on days 1, 57, and 91 for the 2 and 6 mg/(kg day) groups, and on days 1, 57, and 66 for the 20/12 mg/(kg day) group [only a limited number of samples (0, 6, and 24 h) were collected on day 66]. In the 9-month study, full pharmacokinetic profiles were evaluated on day 1 and on days 28, 98, and 274 (last dosing day of treatment cycles 1, 3, and 8, respectively, for the 0.3, 1.5, and 6 mg/(kg day) groups, except that dosing at 6 mg/(kg day) was stopped after completion of the fifth treatment cycle). Single samples were also collected pre-dose on days 36, 71, 106, 141, 176, 211, and 246 (corresponding to treatment cycles 2, 3, 4, 5, 6, 7, and 8, respectively); 4 h post-dose on days 94, 164, and 269 (24th dose of treatment cycles 3, 5, and 8, respectively); and 24 h post-dose on days 64, 134, 169, 204, and 239 (last dosing day of treatment cycles 2, 4, 5, 6, and 7, respectively). Additional samples were collected 1, 4, and 8 weeks after termination of sunitinib treatment. Harvested plasma was stored frozen at −80°C and protected from light until analysis. Other repeat-dose studies were conducted in a similar fashion.
Analysis methods
Sunitinib and SU12662 assay
High-performance liquid chromatography–tandem mass spectrometric (LC/MS–MS) methods were developed and validated for measurement of sunitinib and SU12662 concentrations in rat plasma samples. The samples were prepared for analysis by either ethyl acetate extraction (used in only a few initial studies) or protein precipitation (in test tubes or 96-well plates) and the assay used either propranolol or [2H10]-sunitinib as an internal standard. The concentration range of either 1–2,000 ng/mL or 0.1–200 ng/mL was validated. Calibration and quality control (QC) samples were assayed concomitantly to help ensure acceptable accuracy and precision in the assay results. The analytical method was adequately cross-validated for the assessment of mouse and monkey plasma. Monkey tissue samples harvested during an 8-week study were assayed by modified LC/MS–MS methods involving tissue homogenization and solvent extraction or protein precipitation in sample preparation. Since sunitinib in solution is photosensitive, care was taken during the assays to protect the study samples, calibration and QC, and standard stock solutions from light exposure. A brief description of the LC/MS–MS assay methods for rat plasma follows.
For the test tube method, aliquots of 50 μL of rat calibration standard samples, study plasma samples or QC samples together with 25 μL of internal standard (propranolol) were mixed with 2 mL of acetonitrile in test tubes on a vortex mixer for approximately 1 min. After centrifugation at 4,000 rpm for 5 min, the organic phase was separated and evaporated to dryness at 37°C under nitrogen. The residue was dissolved in 300 μL of a 1:1 mixture of mobile phases A:B. Following vortex mixing and centrifugation, the prepared samples were transferred to injection vials, and aliquots of 5–20 μL were injected into a Keystone BetaBasic-18 column (4.6 × 100 mm, 5 μ) for LC-MS/MS analysis (Shimadzu Series 10 ADVP HPLC system and Perkin-Elmer Sciex API-3000 triple quadrupole mass spectrometer). The gradient mobile phases were 10 mM ammonium acetate in methanol:water (10:90) with 0.1% formic acid (A), and 10 mM ammonium acetate in methanol:water (90:10) with 0.1% formic acid (B). The flow rate was 0.8 mL/min, which was reduced to 0.3 mL/min before introduction into the mass spectrometer. The mass spectrometer was operated using ionization and scan modes of positive ion electrospray and multiple reaction monitoring (MRM), respectively. Each analyte was detected by tandem MS by monitoring specific molecular ion → fragment ion transitions: m/z 399.1 → 325.9 for sunitinib, 371.1 → 283.1 for SU12662, and 360.1 → 116.0 for propranolol.
Calibration curves were constructed by plotting the analyte/internal standard peak area ratios against the analyte concentrations. A weighted linear regression (1/C 2) was used to calculate the concentrations of sunitinib and SU12662 in the study and QC samples. This assay method demonstrated acceptable selectivity/specificity, linearity, and intra- and inter-assay precision and accuracy. The blank matrix also showed no significant interference or carryover on the LC/MS–MS system. The lower limit of quantitation (LLOQ) was 0.1 ng/mL for both sunitinib and SU12662. Sample dilution analysis, freeze-thaw matrix stability, matrix stability at room temperature, processed sample stability, and robustness of the assay were also acceptable.
Alternatively, aliquots of rat plasma (50 μL) were mixed with 500 μL of methanol containing [2H10]-sunitinib as an internal standard in a 96-well polypropylene dual membrane microplate. The methanolic phase was filtered and collected into a second 96-well plate by applying a vacuum to the plate. The methanol was dried under nitrogen gas at 37°C and the residue reconstituted with 200 μL of 15 mM ammonium formate, pH 3.25. After vortex mixing and centrifugation, aliquots of 20 μL of the resulting solutions were injected into the LC–MS/MS system (HP 1100 HPLC system with Perkin-Elmer 200 autosampler and Perkin-Elmer Sciex API-3000 triple quadrupole mass spectrometer). A Symmetry Shield C8 column (2.1 × 50 mm, 3.5 μ) was used to perform the chromatographic separation. The mobile phase was 15 mM ammonium formate buffer solution (pH 3.25):acetonitrile (75:25, v/v), with a flow rate of 0.35 mL/min. The mass spectrometer was operated using MRM (m/z for sunitinib and SU12662 were as described above; m/z 409.1 → 325.9 for internal standard) and in a positive ion mode.
Calibration curves were constructed (as described above) to calculate the concentrations of sunitinib and SU12662 in the study and QC samples. The lower and upper limits of quantitation were 0.07 and 241 ng/mL for sunitinib and 0.07 and 220 ng/mL for SU12662. This method also showed acceptable assay specificity/selectivity, linearity, sensitivity, precision, accuracy, and stability.
Radioactivity assay
Radioactivity in rat and monkey blood, plasma, urine, and fecal samples was measured by liquid scintillation counting (Packard 1900 or 2100 TR Liquid Scintillation Counter). The counting efficiencies were calculated by the external standard method, using a series of quenched standards (supplied by Packard), generating a calibration curve. The counting error was typically <3% for all samples. The background disintegration rate was measured and subtracted from the sample disintegration rate. In general, the limit of quantification was considered to be twice the background level.
Total radioactivity in rat organs/tissues was also measured by quantitative whole body autoradiography (QWBA), via video densitometry of the digital images of the autoradiograms [Fuji BAS 1500 or FLA-5000 image analyzer and AIDA image analysis software (Raytest, Straubenhardt, Germany)].
Pharmacokinetic analysis
Plasma pharmacokinetic parameters of sunitinib and SU12662 were calculated using non-compartmental methods with the aid of WinNonLin (Version 2.0 or greater; Pharsight Inc., Mountain View, CA, USA) or Watson LIMS (Version 2.3 or greater; Thermo Fisher Scientific, Madison, WI, USA). Typical procedures are described below.
After oral dosing, the maximum plasma concentration (C max) and time to maximum concentration (T max) were determined directly from the concentration–time data. Data in the terminal log-linear phase were analyzed by linear regression to estimate the terminal rate constant (k) and half-life (t 1/2 = 0.693/k). At least the last three time points were used to calculate k. Total AUC0–∞ was determined as the sum of AUC through the last measurable concentration (AUC0–t last; calculated using the linear trapezoidal rule) and the terminal area [calculated by dividing the concentration at the last time point (C t last) by k], i.e. AUC0–∞ = AUC0–t last + C t last/k. The linear trapezoidal rule was also used to calculate AUC0–24.
Total plasma clearance (CL) after intravenous dosing was calculated as dose/AUC0–∞, and the volume of distribution at steady-state (V ss) was calculated as CLss/k. Mean AUC0–∞, IV was used for the estimation of oral bioavailability. Total percent bioavailability (%F) was estimated as follows: \( \% F = \left[ {\left( {{\text{AUC}}_{{0 - \infty ,{\text{PO}}}} /{\text{dose}}_{\text{po}} } \right)/\left( {{\text{AUC}}_{{0 - \infty ,{\text{IV}}}} /{\text{dose}}_{\text{iv}} } \right)} \right] \times 100.\)
To describe the pharmacokinetic parameters in the intravenous study in monkeys with plasma sample collection up to 984 h (following a single dose of sunitinib 3 mg/kg), a three-compartmental model with a weight factor of 1/y 2 was used. The contribution to total exposure by the γ terminal phase was calculated using the equation: \( \% {\text{AUC}}\gamma = {\text{C}}/\gamma /\left( {{\text{A}}/\alpha + {\text{B}}/\beta + {\text{C}}/\gamma } \right) \times 100\% . \) Similarly, plasma kinetics of SU12662 were analyzed based on a two-compartmental pharmacokinetic model.
In the repeat-dose studies in rats and monkeys, C max, T max, and AUC of sunitinib and SU12662 over the dosing interval (24 h) were estimated on days when plasma samples were collected to allow for estimation of these parameters. The sum of AUC for sunitinib and SU12662 was utilized for comparison of systemic exposure after repeat-dose administration of sunitinib, because the metabolite exhibits similar potency and pharmacologic activities compared to the parent drug [11].
Results
Single-dose disposition kinetics
Mice
After intravenous administration, sunitinib 1 and 10 mg/kg showed respective systemic clearances of 86 and 68 mL/(min kg), short elimination half-lives of 1.2 and 1.3 h, and volumes of distribution significantly larger than total body water (8.6 and 7.6 L/kg; Table 1). After oral administration, sunitinib (20–160 mg/kg in solution and in suspension formulations) was readily absorbed with a mean T max of 0.5–6.0 h, and a mean t 1/2 of 1.5–7.6 h (Table 1). The mean C max and AUC of sunitinib increased approximately in proportion with dose, and absolute oral bioavailability was moderate (53–77%) and independent of dose (Table 1). Sunitinib and SU12662 appeared to demonstrate higher C max and shorter T max values following an oral dose of sunitinib 40 mg/kg in solution in lactic acid-cremophor cosolvent compared with the same dose in a citrate buffer suspension. However, the half-life and AUC values were not affected by the formulations (Table 1). After an oral dose of sunitinib 40 mg/kg, SU12662 showed a similar T max and half-life to the parent drug, with C max and AUC approximately 60–70% of the parent drug (Table 1). Representative plasma concentration-time profiles for sunitinib following single-dose oral administration of a 40 mg/kg dose are presented in Fig. 1a.

Mean single-dose plasma concentration–time profiles of sunitinib and its major metabolite, SU12662, in mice, rats, and monkeys with oral (a and c) and intravenous (b) dosing. a Plasma concentrations of sunitinib following oral dosing in mice (40 mg/kg) and rats (10 mg/kg). b Plasma concentrations of sunitinib and SU12662 following intravenous dosing in rats (4 mg/kg) and monkeys (3 mg/kg). c Plasma concentrations of sunitinib following oral dosing at 6 mg/kg in male and female monkeys
In vitro studies showed that sunitinib and SU12662 were highly bound (~91 and ~95%, respectively) to mouse plasma proteins and that the fraction bound was independent of concentration (0.25, 1, and 10 μM). In addition, sunitinib and SU12662 were preferentially partitioned into erythrocytes rather than plasma: following an intravenous dose of sunitinib 10 mg/kg, blood to plasma concentration ratios were 1.5–2.3 for sunitinib and 1.3–2.6 for SU12662. Central nervous system (CNS) penetration of sunitinib in mice (dosed intravenously at 9.2 mg/kg) was rapid, and brain concentrations were higher than plasma concentrations up to 1 h after dosing (at 5 and 60 min post- dose, respectively, brain levels were 9.2 and 2.3 nmol/g, and plasma levels were 1.3 and 0.3 nmol/mL).
Rats
Representative plasma concentration–time profiles for sunitinib and SU12662 following single oral (10 mg/kg) and intravenous (4 mg/kg) doses, respectively, are presented in Figs. 1a, b. In a study with intravenous doses of sunitinib 1, 4, or 8 mg/kg, sunitinib had a short elimination half-life of 1.9–2.5 h, plasma clearance of 19–55 mL/(min kg), and a volume of distribution (3.9–9.3 L/kg) that was significantly larger than total body water. Sunitinib showed apparent non-linear kinetics over this dose range, as sunitinib AUC increased greater than dose-proportionally, while the volume of distribution and clearance decreased with dose (Table 2), suggesting saturation of a clearance pathway. There were also apparent gender-related differences in the pharmacokinetics of sunitinib and SU12662 in rats. Female rats had higher dose-normalized sunitinib AUC compared with male rats (for example, after an intravenous dose of sunitinib 4 mg/kg to male rats and 14C-sunitinib 5 mg/kg to female rats, the mean sunitinib AUC was 2,334–3,123 and 7,202 ng h/mL [5,723 ng h/mL when normalized to 4 mg/kg], respectively), while the metabolite AUC was somewhat higher in male rats (6,723 ng h/mL vs. 5,379 ng h/mL in female rats when dose-normalized; Table 2). SU12662 AUC was approximately double the parent drug AUC (SU12662:sunitinib AUC ratio = 2.15) in male rats, while they were similar in female rats (AUC ratio = 0.93).
After oral administration of 2, 10, or 20 mg/kg in male rats, sunitinib had a T max of 3.0–6.0 h and a half-life of approximately 2–3 h (Table 2). The AUC of sunitinib increased with dose (Table 2). Low plasma concentrations of sunitinib after the 2 mg/kg dose permitted the estimation of AUC0–t last only. Non-linear kinetics were not observed from 10 to 20 mg/kg. With sunitinib AUC following the intravenous dose of 8 mg/kg as a reference, the bioavailability of sunitinib was 55 and 57% in the male rats with doses of 10 and 20 mg/kg, respectively. Consistent pharmacokinetic parameters were observed in a second study with an oral dose of 15 mg/kg (Table 2). In another study with [14C]-sunitinib (15 mg/kg given orally), sunitinib AUC in male rats was approximately 50% of that in female rats, while SU12662 AUC in male rats was almost double that in female rats (Table 2). The SU12662:sunitinib AUC ratio was 5.81 and 1.55 in male and female rats, respectively. In female rats, with the intravenous dose of 5 mg/kg as a reference, the absolute bioavailability of sunitinib was 111% for an oral dose of 15 mg/kg.
As in mice, in vitro studies in rats showed that sunitinib and SU12662 were highly plasma protein bound (approximately 98% for both) and the fraction bound was independent of drug concentration (0.25, 1, and 10 µM). The erythrocyte uptake of sunitinib and SU12662 in rats was unclear due to inconsistent ex vivo and in vitro data. The available data indicated that sunitinib and SU12662 were either equally distributed between plasma and erythrocytes or had preferential uptake into erythrocytes. Quantitative whole-body autoradiography in rats showed that drug-related radioactivity was extensively distributed in tissues. At 3, 6, and 24 h after dosing, all tissues had higher (up to 70-fold higher) concentrations of radioactivity compared with blood; the only exceptions were brain, spinal cord, and white fat, with lower or similar concentrations compared with blood. Male and female albino and pigmented rats demonstrated similar patterns of absorption, distribution, and elimination of drug-related radioactivity—except for the pigmented tissues in the eye (including uveal tract) and skin of pigmented rats.
Drug-related radioactivity in rats was excreted mainly in feces, with urinary excretion a minor route. The amounts of radioactivity recovered (as a % of the dose) over 72 h in urine and feces, respectively, were 9.1 and 77.2% for female rats after intravenous dosing, 9.3 and 71.1% for female rats after oral dosing, and 8.5 and 75.2% for male rats after oral dosing. The mean total recovery across all groups was approximately 82–87%. In a study in bile-duct cannulated rats, drug-related radioactivity was excreted in bile (43.0 and 39.2% of a 15 mg/kg oral dose were excreted in bile and feces, respectively, with a total recovery of 96.1% in 48 h) but enterohepatic recirculation was not observed. Drug-related radioactivity was also secreted in the milk, with concentrations ≥5-fold higher than in plasma at 3–24 h after an oral dose of 15 mg/kg to lactating rats.
Monkeys
Representative plasma concentration–time profiles for sunitinib and SU12662 following single intravenous (3 mg/kg) and oral (6 mg/kg) doses of sunitinib, respectively, are shown in Fig. 1b and c; pharmacokinetic parameters are summarized in Table 3. Despite differences in study design, sunitinib clearance [25.0–35.7 mL/(min kg)], volume of distribution (13.3–21.1 L/kg), and absolute bioavailability (41–58%) were fairly consistent across studies. However, the terminal-phase half-life was dependent on the length of the plasma sample collection period following dose administration. For example, a prolonged terminal-phase half-life was estimated in a study with sampling times extended up to 984 h (Table 3). In this study, the concentrations of sunitinib and SU12662 decreased with time tri- and bi-exponentially, respectively. The relatively consistent clearance and volume of distribution across studies suggested that the prolonged terminal-phase half-life did not contribute significantly to the total AUC, and compartmental pharmacokinetic analysis indicated that the terminal-phase AUC accounted for ≤1% of the total AUC. Therefore, the moderate β-phase half-life of sunitinib (14.9 h) and α-phase half-life of SU12662 (15.7 h) were considered to be the effective half-lives. The limited-to-moderate degree of accumulation after repeat-dose administration (see “Repeat-dose data”) was consistent with the moderate effective half-lives. After intravenous administration of sunitinib, the AUC of SU12662 was 28–40% of the AUC of the parent drug, while these values increased to 91–116% after oral administration of sunitinib. Similar to rats, the increased plasma concentrations of SU12662 after oral administration of sunitinib compared with intravenous dosing suggested some degree of first-pass metabolism in monkeys. There were no gender-related differences in the pharmacokinetics of sunitinib and SU12662 in monkeys.
In vitro studies in monkeys showed that sunitinib and SU12662 were highly plasma protein bound (~95 and 86%, respectively) and that the fraction bound was independent of drug concentrations (0.25, 1, and 10 µM). Additionally, sunitinib was preferentially partitioned into erythrocytes rather than plasma (blood to plasma concentration ratios were 1.7–1.9 for sunitinib and 2.9–3.1 for SU12662; K p of total radioactivity was approximately 2–5, and in vitro mean K p was 4.1 for sunitinib).
Similar to results observed for the rat, drug-related radioactivity in monkeys was excreted mainly in feces; urinary excretion was a minor route. The amounts of radioactivity recovered (as a % of the oral dose) over 336 h in urine and feces, respectively, were 6.1 and 84.1% (for female monkeys) and 4.8 and 87.3% (for male monkeys). The mean total recovery across all groups was approximately 90–94%.
Repeat-dose data (Toxicokinetics)
Mice
Following daily oral dosing of sunitinib to SKH1 mice for up to 5 weeks, plasma concentrations of sunitinib and SU12662 increased proportionally with the increase in dose from 40 to 80 mg/(kg day) (data not shown). Plasma concentrations appeared to reach steady-state by day 7. Data comparisons showed no significant differences in exposure to sunitinib between days.
Rats
Both sunitinib and SU12662 were evaluated in the 3- and 6-month repeat-dose studies. In these studies, sunitinib was readily absorbed and steadily eliminated to afford adequate systemic exposure following once-daily dosing.
In the 3-month study, male and female rats were dosed with 1.5 or 5 mg/(kg day) for 91 days or with 15 mg/(kg day) for 65 days. Pharmacokinetic parameters—which were evaluated on days 1, 28, 65 [15 mg/(kg day) group only], and 91—are summarized in Table 4. [An extended and comprehensive version of Table 4 (Supplementary Table 4a) presenting data from animals treated with sunitinib for 28 days, 3 and 6 months is available as supplementary data in the online journal.] As a representative of daily exposure at steady-state, the concentration-time profiles for sunitinib and SU12662 on day 28 are shown in Fig. 2a, b, respectively. Since sunitinib and SU12662 have shown similar activity in in vitro biochemical assays [11], the total exposure—measured by the sum of AUC for sunitinib and SU12662—was considered more relevant for the evaluation of toxicologic effects than the exposure of sunitinib or SU12662 alone; Fig. 3a shows the dose-total-exposure relationships at steady-state. The mean C max and AUC of sunitinib and SU12662 increased greater than dose-proportionally from 1.5 to 15 mg/(kg day) (Table 4). The increases in C max and AUC from days 1 to 28 were ≤3-fold. Both male and female rats showed higher exposure (1.5- to 4-fold) to SU12662 than to sunitinib, and the exposure to sunitinib in the female rats was higher than in the males. Although the AUC and C max of both sunitinib and SU12662 appeared to continue to increase from days 1 to 91, there were no increases in exposure from days 28 to 65 for the 15 mg/(kg day) groups, and for the 1.5 and 5 mg/(kg day) groups the increases in exposure from days 28 to 91 were less than 2-fold. These results suggested that the systemic exposure was approaching steady-state by day 28.

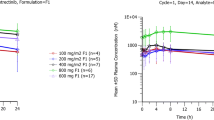
Steady-state plasma concentration–time profiles of sunitinib and SU12662 in rats on day 28 of a 3-month study (a and b) and in monkeys on day 57 of a 3-month study (c and d) (in these figures, ‘m’ represents male animals and ‘f’ represents female animals). a Sunitinib (3-month rat study). b SU12662 (3-month rat study). c Sunitinib (3-month monkey study). d SU12662 (3-month monkey study)

Steady-state total exposure (sum of AUC of sunitinib and SU12662) versus dose in rats on day 28 of a 3-month study (a) and in monkeys on day 57 of a 3-month study (b); a 3-month rat study, b 3-month monkey study
The 6-month study was conducted with sunitinib 0.3, 1.5, or 6 mg/(kg day) administered in five cycles according to a 4/1 schedule. Sunitinib and SU12662 mean C max and AUC generally increased with dose, with exceptions observed on day 28 when some exposure values exhibited high variability (data not shown). Although female rats tended to have higher exposure to sunitinib and lower exposure to SU12662 compared with male rats, the gender-related differences were not consistent across all treatment groups and days. The treatment-day effects were confounded by the high variability of data on day 28. Comparison of C max and AUC on days 168 and 1 indicated that exposure to sunitinib and SU12662 increased with repeated-dose administration: days 168 to 1 ratios for C max and AUC were in the approximate range of 2–5 and 2–4, respectively. However, the plasma concentrations of sunitinib and SU12662 in pre-dose samples from the second to fifth treatment cycles (on days 35, 70, 105, and 140, respectively) were below the LLOQ, with a few exceptions near the LLOQ. These results suggested that steady-state exposure to sunitinib and SU12662 was attained by the end of the first treatment cycle. The drug and metabolite plasma concentrations were also below the LLOQ during the final recovery period on days 175, 196, and 224 (1, 4, and 8 weeks post last dose administration).
Monkeys
Both sunitinib and SU12262 were evaluated in the 3- and 9-month repeat-dose studies in monkeys. In these studies, sunitinib was readily absorbed and steadily eliminated after once-daily oral doses, providing adequate and sustained systemic exposure. As a representative of repeat-dose exposure in monkeys, plasma concentration–time profiles for sunitinib and SU12662 on day 57 of the 3-month study are shown in Fig. 2c and d; Fig. 3b shows the sunitinib + SU12662 AUC versus dose at steady-state.
In the 3-month study, the once-daily dose started at 2, 6, or 20 mg/kg. Due to intolerance, the 20 mg/(kg day) dose was reduced to 12 mg/(kg day) on day 29; the monkeys in this dose group were given a drug-free period from days 34 to 42 and eventually removed from dosing on day 67. In this study, C max and AUC of sunitinib were higher than those of SU12662, and the sunitinib:SU12662 AUC ratio did not change markedly with increasing dose or with multiple dosing (Table 5). [An extended and comprehensive version of Table 5 (Supplementary Table 5a) presenting data from animals treated with sunitinib for 28 days, 8 weeks, 3 and 9 months is available as supplementary data in the online journal.] Across all dose groups and collection days, there were no marked gender differences in C max and AUC. The increases in systemic exposure were proportional to dose from 2 to 6 mg/(kg day), but the increases were greater than dose-proportional at higher doses. The C max and AUC of sunitinib and SU12662 on days 57 and 91 were generally higher than on day 1, indicating accumulation of drug and metabolite following multiple-dose administration of sunitinib. Days 57 to 1 ratios of C max and AUC for sunitinib and SU12662 in both male and female monkeys were in the approximate range of 1–3; days 91/66 to 1 ratios were in the approximate range of 2–5 (with dose normalization where applicable).
The 9-month study was conducted in eight cycles of once-daily dosing with sunitinib 0.3, 1.5, and 6 mg/kg according to a 4/1 schedule. Gender-related differences in exposure were not apparent, and sunitinib exposure was generally greater than or equal to SU12662 exposure (data not shown). Sunitinib and SU12662 C max and AUC generally increased with dose, but varied across treatment cycles. Exposure-treatment day effects were not consistent, as sunitinib and SU12662 C max and AUC fluctuated across the profiling days. The reasons for these observations are not known. However, comparison of 0- and 24-h plasma concentrations of sunitinib and SU12662 on different treatment days and cycles showed that these trough concentrations were similar and did not consistently increase with dosing cycles. These results indicated that, despite the apparent variability of plasma concentration data on different treatment days, the animals in this study were exposed to sunitinib and its metabolite, with the target exposure (plasma concentrations >50 ng/mL) likely attained by the end of the first dosing cycle.
Tissue distribution. In another repeat-dose monkey study in which daily oral doses of sunitinib 6 and 12 mg/kg were administered for 8 weeks or for two cycles of 4 weeks (4/2 regimen), the total drug (sunitinib plus SU12662) concentrations in most tissues were 13- to 308-fold higher than plasma concentrations at 24 h after the last dose for both dose groups. White fat concentrations were 2- to 14-fold higher than in plasma, while the ratios in brain were close to unity (1- to 3-fold higher). However, the tissue concentrations had attained steady-state by the fourth week of daily dosing and there was no continuous accumulation of sunitinib or SU12662 in the tissues from day 28 to 56 or from the first to second 4/2 dosing cycles. Therefore—similar to rats, and consistent with the high volume of distribution observed in single-dose monkey studies (Table 3)—sunitinib and SU12662 were extensively distributed in tissues in monkeys, with most tissues attaining higher concentrations than plasma and reaching steady-state by 28 days of dosing. Upon termination of drug treatment, the tissue concentrations decreased with time, with no tissues examined showing prolonged retention of high concentrations of the drug or metabolite.
Discussion
Sunitinib is an oral multitargeted tyrosine kinase inhibitor that has shown clinical activity in advanced RCC and in GIST after disease progression on, or intolerance to, imatinib. To support the clinical development of sunitinib, extensive non-clinical studies of the pharmacokinetics and dispositional properties of sunitinib and its primary active metabolite (SU12662) were performed as reported here. These data indicate that sunitinib exhibits advantageous pharmacokinetic and dispositional properties required for an oral therapeutic agent in humans; namely, linear kinetics at clinically relevant doses, rapid and extensive absorption, a long half-life enabling once-daily dosing, and a large volume of distribution without metabolic induction suggesting that clinically relevant (i.e. therapeutic) concentrations can be achieved.
The pharmacokinetics of sunitinib and SU12662 after oral sunitinib administration were generally consistent with those observed after intravenous administration. The gender-related differences apparent in rats after oral and intravenous administration were probably due to the fact that male rats are more extensive CYP3A-mediated metabolizers than female rats [18]. Such gender-related differences were specific to and relevant for rats only [11]. The proper estimation of sunitinib oral bioavailability in rats was confounded by the non-linear and gender-dependent kinetics, and differences between the intravenous (reference) and oral (test) doses. Nonetheless, it can be concluded that the oral bioavailability of sunitinib in mice, rats, and monkeys was ≥50%.
Following oral administration of sunitinib, T max of SU12662 was slightly later than or similar to T max of the parent drug, indicating a rapid first-pass effect. Also, in both rats and monkeys, the metabolite:parent AUC ratios were higher after oral compared with intravenous dosing, indicating that sunitinib may be subject to first-pass metabolism on absorption in animals [12].
Both sunitinib and drug-related materials, including SU12662, were extensively bound to plasma proteins (86–98%) in the non-clinical species studied, and showed preferential uptake into red blood cells from plasma in mice and monkeys. After intravenous sunitinib administration in mice, rats, and monkeys, sunitinib and SU12662 had volumes of distribution significantly larger than total body water (3–21 L/kg).
When tissue concentrations of sunitinib and SU12662 were examined, it was clear that both compounds were widely distributed into tissues, including the CNS [19], which, in mice, showed higher concentrations than plasma compared with monkeys and rats in which CNS concentrations were similar to or lower than in plasma. Otherwise, most tissues had much higher concentrations of drug compared with blood, consistent with a high volume of distribution. Of note, pigmented tissues in the eye (including the uveal tract) and skin of pigmented rats showed longer retention of higher drug levels than non-pigmented tissues. This was believed to be associated with melanin binding, which occurs benignly with many basic drugs [20].
After intravenous sunitinib administration, sunitinib and SU12662 exhibited moderate-to-high systemic clearances in all species. In rats and monkeys, orally and intravenously administered doses were quantitatively recovered (as drug and drug-related materials) mainly in feces (71–77% of the dose in rats; 84–87% of the dose in monkeys), with urinary excretion forming a minor route of elimination (approximately 9% of the dose in rats; 5–6% of the dose in monkeys). In rats, 43% of the dose was excreted in bile, but there were no indications of enterohepatic recirculation of drug-related materials. Drug-related radioactivity was also excreted in the milk of lactating rats.
Data from the current studies in mice support the findings of previous murine studies reported by Mendel et al. [2], who demonstrated that sunitinib has predictable, dose-dependent pharmacokinetics with dose-dependent changes in exposure. Mendel and colleagues showed that efficacy is driven by maintenance of the target plasma concentration; thus, effective exposure is achieved with plasma sunitinib concentrations maintained above 50 ng/mL for 12 h with daily dosing, corresponding to an administered dose of 40 mg/(kg day).
The repeat-dose pharmacokinetics of sunitinib and SU12662 in rats and monkeys (the primary animal species for non-clinical safety evaluations) were consistent with the kinetics after single doses. A few differences, however, were noted: unlike in rats, there was no gender-related difference in metabolism of sunitinib to SU12662 in monkeys. Quantitatively, after repeat-dose administration of sunitinib, again, unlike in rats, the SU12662 AUC in monkeys was generally smaller or similar compared with the AUC of the parent drug. Similarly, in humans, upon repeat dosing, the metabolite accounts for 23–37% of the total drug AUC [11].
The plasma half-life of sunitinib allowed once-daily dosing in rats and monkeys to attain adequate systemic exposure in repeat-dose studies. Systemic exposure generally increased greater than dose-proportionally, and attained steady-state by 4 weeks of daily dosing.
Consistent with the long half-lives, there was limited-to-moderate accumulation of sunitinib and SU12662 in plasma following repeat-dose administration in rats and monkeys with a ≤3-fold observed increase in sunitinib and SU12662 systemic exposure through the last day of dosing. The long terminal-phase half-lives observed in monkeys when plasma concentrations were monitored for a prolonged period did not lead to significant drug accumulation following repeated administration and contributed to <1% of the AUC. This indicates that a continuous dosing regimen would not be expected to result in significantly higher exposures over time, as compared to a cyclical regimen. The predictable and dose-dependent pharmacokinetics of sunitinib were key factors supporting the continued assessment and development of the compound [2].
In summary, in the non-clinical species studied, sunitinib exhibited advantageous oral pharmacokinetic and dispositional properties: linear kinetics at clinically relevant doses; rapid and extensive absorption; long half-lives enabling once-daily dosing; and a large volume of distribution without metabolic induction, suggesting that clinically relevant (i.e. therapeutic) concentrations can be achieved. In the range of doses expected to lead to clinically relevant exposure in humans, C max and AUC generally increased proportionally with dose, allowing for the extrapolation of pharmacokinetic and toxicokinetic data for use in safety margin assessment and tumor xenograph mouse models for efficacy assessment [2–4, 16]. The data collected from the single- and repeat-dose studies in mice, rats, and monkeys performed as part of the sunitinib non-clinical development program facilitated the implementation and conduct of clinical studies, which subsequently led to regulatory approval of sunitinib.
Abbreviations
- AUC0–∞ :
-
Total area under the concentration–time curve from time zero to infinity
- AUC0–t last :
-
AUC from time zero to the last measurable concentration
- AUC0–24 :
-
AUC from time zero to 24 h
- CLplasma :
-
Total plasma clearance
- C max :
-
Maximum plasma concentration
- %F :
-
Total percent bioavailability
- FLT3:
-
Fms-like tyrosine kinase-3 receptor
- GIST:
-
Gastrointestinal stromal tumors
- K :
-
Terminal rate constant
- KIT:
-
Stem cell factor receptor
- K p :
-
Red blood cell to plasma radioactivity partitioning ratio
- LC–MS/MS:
-
High-performance liquid chromatography–tandem mass spectrometry
- MRM:
-
Multiple reaction monitoring
- NF:
-
National Formulary
- PDGFR:
-
Platelet-derived growth factor receptor
- QWBA:
-
Quantitative whole body autoradiography
- RET:
-
Glial cell-line derived neurotrophic factor receptor (REarranged during Transfection)
- RTK:
-
Receptor tyrosine kinase
- t 1/2 :
-
Half-life
- T max :
-
Time to maximum concentration
- USP:
-
US Pharmacopeia
- VEGFR:
-
Vascular endothelial growth factor receptor
- V ss :
-
Volume of distribution at steady-state
References
Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM (2003) SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther 2:471–478
Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM (2003) In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 9:327–337
Murray LJ, Abrams TJ, Long KR, Ngai TJ, Olson LM, Hong W, Keast PK, Brassard JA, O’Farrell AM, Cherrington JM, Pryer NK (2003) SU11248 inhibits tumor growth and CSF-1R-dependent osteolysis in an experimental breast cancer bone metastasis model. Clin Exp Metastasis 20:757–766
O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, Heinrich MC, Cherrington JM (2003) SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 101:3597–3605
Brahmer JR, Govindan R, Novello S, Rosell R, Belani CP, Atkins JN, Gillenwater HH, Tye L, Chao R, Socinski MA (2007) Efficacy and safety of continuous daily sunitinib dosing in previously treated advanced non-small cell lung cancer (NSCLC): results from a phase II study. Proc Am Soc Clin Oncol 25:7542
Kulke MH, Lenz H-J, Meropol NJ, Posey J, Picus J, Ryan DP, Bergsland E, Stuart K, Baum CM, Fuchs CS (2005) Results of a phase II study with sunitinib malate (SU11248) in patients (pts) with advanced neuroendocrine tumors (NETS) (oral presentation). In: The 13th European cancer conference (ECCO 13), Paris, October 30–November 3, Abstract 718
Saltz LB, Rosen LS, Marshall JL, Belt RJ, Hurwitz HI, Eckhardt SG, Bergsland EK, Haller DG, Lockhart AC, Rocha Lima CM, Huang X, DePrimo SE, Chow Maneval E, Chao RC, Lenz HJ (2007) Phase II trial of sunitinib in patients with metastatic colorectal cancer after failure of standard therapy. J Clin Oncol 25:4793–4799
Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, Lehman M, Adams BJ, Bello CL, DePrimo SE, Baum CM, Miller KD (2008) A phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol 26:1810–1816
Socinski MA, Novello S, Brahmer JR, Rosell R, Sanchez JM, Belani CP, Govindan R, Atkins JN, Gillenwater HH, Palleres C, Tye L, Selaru P, Chao RC, Scagliotti GV (2008) Multicenter, phase II trial of sunitinib in previously treated, advanced non-small-cell lung cancer. J Clin Oncol 26:650–656
Zurita AJ, Shore ND, Kozloff MF, Ryan CW, Beer TM, Maneval EC, Chen I, Logothetis CJ (2007) Distinct patterns of PSA modulation by single-agent sunitinib before combination with docetaxel and prednisone in patients with metastatic castrate-resistant prostate cancer (CRPCa). Proc Am Soc Clin Oncol 25:5134
Pfizer Inc. (2008) SUTENT® (sunitinib malate) capsules, oral. Prescribing information (revised: May 2008). http://www.sutent.com
Washington C, Eli M, Bello C, Schaaf L, Polasek E, Tan LH, Scigalla P, Sarapa N (2003) The effect of ketoconazole (KETO), a potent CYP3A4 inhibitor, on SU011248 pharmacokinetics (PK) in Caucasian and Asian healthy subjects. Proc Am Soc Clin Oncol 22:138 (Abstract 553)
Bello C, Houk B, Sherman L, Misbah S, Sarapa N, Smeraglia J, Haung X (2005) Effect of rifampin on the pharmacokinetics of SU11248 in healthy volunteers. Proc Am Soc Clin Oncol 23:3078
Bello C, Peng G, Patyna S, Poole W, Smeraglia J, Sherman L, Garrett M, Klamerus K (2007) A phase I mass-balance study to evaluate the metabolism and excretion of [14C]-sunitinib (SU11248) in healthy male subjects (abstract). In: American association for cancer research annual meeting, Los Angeles, April 14–18, Abstract LB-354
Bello CL, Sherman L, Zhou J, Verkh L, Smeraglia J, Mount J, Klamerus KJ (2006) Effect of food on the pharmacokinetics of sunitinib malate (SU11248), a multi-targeted receptor tyrosine kinase inhibitor: results from a phase I study in healthy subjects. Anticancer Drugs 17:353–358
Khosravan R, Toh M, LaFargue J, Ni G, Bello C (2008) Sunitinib pharmacokinetic (PK) and safety data in subjects with renal impairment and on hemodialysis. J Clin Oncol 26 (Abstract 2578)
Patyna S, Heward JK, Evering W (2006) Non-clinical safety evaluation of sunitinib, a novel multitargeted tyrosine kinase inhibitor. Tox Path 35:179–197 (Abstract 42)
Mahnke A, Strotkamp D, Roos PH, Hanstein WG, Chabot GG, Nef P (1997) Expression and inducibility of cytochrome P450 3A9 (CYP3A9) and other members of the CYP3A subfamily in rat liver. Arch Biochem Biophys 337:62–68
Patyna S, Peng G (2006) Distribution of sunitinib and its active metabolite in brain and spinal cord tissue following oral or intravenous administration in rodents and monkeys. Eur J Cancer 4:21 (Abstract 56)
Zane PA, Brindle SD, Gause DO, O’Buck AJ, Raghavan PR, Tripp SL (1990) Physicochemical factors associated with binding and retention of compounds in ocular melanin of rats: correlations using data from whole-body autoradiography and molecular modeling for multiple linear regression analyses. Pharm Res 7:935–941
Acknowledgments
Pfizer Inc. provided financial support for this study. Editorial assistance was provided by ACUMED® (Tytherington, UK) and was funded by Pfizer Inc.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Haznedar, J.Ö., Patyna, S., Bello, C.L. et al. Single- and multiple-dose disposition kinetics of sunitinib malate, a multitargeted receptor tyrosine kinase inhibitor: comparative plasma kinetics in non-clinical species. Cancer Chemother Pharmacol 64, 691–706 (2009). https://doi.org/10.1007/s00280-008-0917-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-008-0917-1