
Abstract
Deep tissue injury (DTI) is a severe kind of pressure ulcers formed by sustained deformation of muscle tissues over bony prominences. As a major clinical issue, DTI affects people with physical disabilities, and is obviously related to the load-bearing capacity of muscle cells in various in vivo conditions. It has been hypothesized that oxidative stress, either induced by reperfusion immediately following tissue unloading or in chronic inflammatory conditions, may affect the cellular capacity against subsequent mechanical damages. In this study, we measured the compressive damage threshold of C2C12 mouse myoblasts with or without pre-treatment of hydrogen peroxide as an oxidative agent to understand how changes in the oxidative environment may contribute to the development of DTI. Spherical indentation was applied onto a layer of agarose gel (3 mm thick) covering a monolayer of C2C12 myoblasts. Cell damage was recognized by using a cell membrane damage assay, propidium iodide. The spatial profile of the measured percentage cell damage was correlated with the radially varying stress field as determined by finite element analysis to estimate the compressive stress threshold for cell damage. Results supported the hypothesis that chronic exposure to high-dosage oxidative stress could compromise the capability of muscle cells to withstand compressive damages, while short exposure to low-dosage oxidative stress could enhance such capability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
People with spinal cord injury (SCI) are likely to suffer from pressure ulcers.4,5,8 Frail patients who are confined to beds are similarly vulnerable.1,2 There are two forms of pressure ulcers, superficial damage and deep tissue injury (DTI).4,22 DTI is a particularly severe kind of pressure ulcers originated in deep tissues around skeletal prominences caused by excessive internal stresses and strains as results of skeletal and epidermal loadings.25,33 As deep, they are hard to be detected clinically at the beginning, but can propagate to cause full-thickness tissue necrosis under repetitive loadings.19,39 The intensity and duration of the loading pressure as well as the tissue tolerance are the main biomechanical factors of pressure ulcers.11 Ischemia, direct mechanical damage, reperfusion oxidative damage, and repair are all coupled with each other under repetitive loading and unloading.21
The mechanism of muscle cell damage and repair is essential to the understanding of DTI.3,32,34,36 Cytoskeleton plays significant roles in the biostructural behaviors of cells and their mechanotransduction mechanisms.28,38 If cell damage is contained within a certain threshold, the cell may be able to repair itself. But if the damage exceeds that threshold, the cell may go down the paths of apoptosis or necrosis,14,18,30 leading to cell death and tissue failure. Compressive damage of cells has been studied on skeletal muscle in the context of DTI.6,14,15,26 The compressive damage stress threshold depends on the compression duration. Such relationship could be described by an inverse sigmoid curve, indicating a sharp decrease in the loading threshold at about 2 h of loading duration.17 The relationship between strain threshold and tolerance duration also showed a similar inverse sigmoid relationship in a tissue-engineered model.15
Oxidative stresses arise from many origins in vivo. It has been reported that oxidative stress accompanies inflammation, aging, neurodegenerative and cardiovascular diseases.29 Oxidative stress can affect myoblast cytoskeleton and induce cell apoptosis.31,35 Both mechanical trauma and prolonged ischemia could increase the permeability of the plasma membrane for Ca2+, and could lead to an increased production of reactive oxygen species.16,23 Chronic inflammation in vivo could also be associated with chronic oxidative stress.13 It has been remarked that post-ischemic reperfusion could lead to oxidative surges9 and thus has also been cited as a factor in the formation of pressure ulcers.27 There have been few literatures on how oxidative stress may affect quantitatively the load-carrying capacity of muscle cells. Different dosages and exposure durations of oxidative stress may have different effects on cells. We hypothesized that oxidative stress might compromise the capability of muscle cells to withstand compressive damages.
Materials and Methods
Cell Culture and Oxidative Treatment
C2C12 mouse skeletal myoblast line (ATCC, Manassas, USA) was grown in monolayer culture in 35 mm diameter tissue culture dishes (SPL Lifesciences, Korea) with growth medium in a 37 °C, 5% CO2 humidified incubator. The growth medium consisted of high-glucose Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Life Technologies, USA), 10% fetal bovine serum (FBS, Gibco, Life Techologies, USA) and 1% Penicillin–Streptomycin (Gibco, Life Technologies, USA). The myoblasts were trypsinized by 0.25% Trypsin–EDTA (1×, Gibco, Life Technologies) for the cell passage process every 3 days to maintain the continuous passage. The myoblasts in this study were of passage 16–19. The monolayers were almost confluent at the time of the experiments. H2O2 was prepared in concentrations of 0.1 and 0.5 mM in cell culture medium.30,35 The myoblasts were respectively immersed in 0.1 mM H2O2 for 1 h (short), 0.5 mM H2O2 for 1 h (short), 0.1 mM H2O2 for 20 h (chronic), and 0.5 mM H2O2 for 20 h (chronic) as the treated samples. After the treatment, Dulbecco’s phosphate-buffered saline (PBS, 1×, Gibco, Life Technologies) was used to wash away the residual H2O2 in the culture dishes for 3 times.
Agarose Hydrogel Fabrication
0.5% (w/v) agarose (Biosharp, Hefei, China) was dissolved in PBS at 100 °C and then cooled in room condition. When the temperature of the agarose solution almost reached 37 °C, it was dispensed into the culture dish to form a 3 mm thick gel layer over a monolayer of myoblasts. One milliliter culture medium was added after the gel was formed.
Compression Device
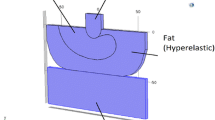
The gel-covered myoblasts monolayer was loaded with compressive stress in a Mach-1™ micromechanical system (Biomomentum Inc). Indentation was conducted right after the gel formation with a half-spherical indenter of 6.38 mm diameter at a rate of 0.01 mm s−1 until the overall deformation at the center reached ~50% (Fig. 1). Such indentation provided a radially varying compressive stress field at the bottom of the gel layer—one that was highest at the center and decreasing along the radius. The Mach-1™ system was housed in an incubator at 37 °C with 5% CO2 throughout the indentation experiments. The half-spherical indenter was connected to a 70 N load cell. The constant strain was maintained for a period from 15 to 300 min as listed in Table 1. The effect of the mere presence of the gel on cell damage was assessed by comparing the percentage of cell damage among three groups of samples: (I) samples not covered by gel and without indentation, (II) samples covered by gel but without indentation; (III) samples covered by gel and with indentation.

Real loading apparatus of agarose gel indentation was conducted at 0.01 mm s−1 speed with a 6.38 mm-diameter stainless steel indenter. The final strain at the center of the gel was about 50%. Myoblasts were attached on the bottom of dish straightly under the agarose gel
Mechanical Property of Agarose Hydrogel
Uniaxial unconfined compression was conducted in the Mach-1™ on the 0.5% agarose gel (3 mm thick and 14.8 mm in diameter) with a flat platen of diameter 25.4 mm to determine the mechanical property of the gel. A 70 N load cell was connected to the indenter. Nominal equilibrium stresses were recorded at points of 10, 20, 30, 40, and 50% nominal strains. The equilibrium stress–strain relationship was analyzed using an Ogden hyperelastic model of the following form:
where U is the strain energy per unit of reference volume; \(\bar{\lambda }_{i}\) are the deviatoric principal stretches \(\bar{\lambda }_{i} = J^{{ - \frac{1}{3}}} \lambda_{i}\); \(\lambda_{i}\) are the principal stretches; J is the total volume ratio; \(J^{\text{el}}\) is the elastic volume ratio; N is a material parameter; \(\mu_{i}\), \(\alpha_{i}\) and \(D_{i}\) are temperature-dependent material parameters.
The stress relaxation after 0.01 mm s−1 ramp up to 30% strain was measured to assess the viscoelastic property of the gel. The subsequent relaxation curve was recorded for 10 min. The shear relaxation modulus can be written in the dimensionless form:
where \(G_{\text{R}} (t)\) is the shear stress relaxation modulus, G 0 = \(G_{\text{R}} (0)\). Viscoelasticity of the gel was defined by a Prony series expansion of the dimensionless relaxation modulus:
where N, \(\bar{g}_{i}^{\text{P}}\) and \(\tau_{i}^{\text{G}}\), i = 1, 2, …, N, are material constants, where \(g_{R} (0) = 1\) and \(g_{\text{R}} (\infty ) = G_{\infty } /G_{0}\). A one-term Prony series was used to describe the viscoelastic property of the hyper-viscoelastic agarose gel. Poisson’s ratio was estimated by a photographic method to measure the transverse strains at various axial strains. Shear modulus was obtained from Poisson’s ratio and Young’s modulus assuming isotropic elasticity.
Finite Element Method Simulation
ABAQUS 6.12 (Simulia, The Netherlands) was used to estimate the profile of the compressive stress acting on the monolayer of myoblasts at the bottom of the agarose hydrogel. A first-order Ogden hyperelastic model24 and a one-term Prony series were assumed in this study. There are many other sophisticated approaches for nonlinear viscoelasticity.10,12 to account for large deformation. In ABAQUS, we have used the visco analysis step with geometric nonlinearity settings.
The elastic modulus and Poisson’s ratio of the indenter were assumed to be 200 GPa and 0.3, respectively. The interface between the indenter and the gel was assumed to be frictionless. The model was meshed with 30,936 8-node linear brick, reduced integration, hourglass control elements (C3D8R). A mesh convergence analysis was performed to ascertain the adequacy of the finite element mesh.
Assessment of Cell Damage
The spatial profiles of cell damage were assessed after indentation for various durations. Propidium iodide (PI) (Sigma, USA) was used as a biomarker for cell damage assessment.7 PI is a fluorescence dye that can penetrate through the cell membrane of damaged cells and combine with DNA and RNA to show red fluorescence. PI cannot stain healthy cells with intact cell membrane. The fluorescence excitation maximum of PI was at 535 nm with the emission maximum at 617 nm. After each specific period of indentation shown in Table 1, 1 mL of PI (10 μg mL−1) was added into the culture dish (n = 3 with power 0.8 while controlling type I error at 0.05). Fluorescence microscopy (Inverted Research Microscope ECLIPSE Ti, Nikon) with G-2B filter was used to capture the red fluorescence of PI. Pictures of the fluorescence field were taken using a CCD camera (DS-Fi1c-L3, Nikon Digital Sight) under a 4× microscope objective on a motorized stage. The pictures were stitched to form one full-field picture of the C2C12 monolayer. The profile of the fluorescence dots and the radially varying stress field under the indented gel were correlated to determine the damage threshold of myoblasts. To estimate the total number of cells in each dish, after fixation with 4% paraformaldehyde solution for 20 min at 4 °C, 1 mL 0.1% Triton X-100 was added for 10 min at the end of the experiment to permeabilize all cell membrane so that all the cells in the dish could be stained by fluorescent PI for counting. The percentage of cells damaged was determined by dividing the number of cells damaged in the indention by the total number of cells.
Imaging Processing
The pictures with PI spots were processed in the ImageJ 1.46r (NIH) into 8-bit pictures. A treated picture was read into Matlab R2010a (Mathworks) for further processing, rendering the grey value of background as 0 and PI spots as 1 by setting a grey value threshold. The grey value threshold was determined by sampling ten randomly selected PI spots. To smooth out the fluorescence image, a 150 × 150 kernel matrix with all elements being one was applied to perform convolution on the picture matrix. The resulted matrix showing the density of the PI spots along with the total cell density provided a spatial profile of percentage cell damage.
Data Analysis
To determine the compression damage threshold for a particular indentation duration, a damage threshold circle was defined for that indentation duration using the criteria that the percentage cell damage within the circle was at least three times that in the peripheral region where the compressive stress should be trivial. The radius of the damage threshold circle at the particular indentation duration was correlated with the radial stress profile obtained from the ABAQUS finite element analysis. A relation between the compressive damage threshold and indentation duration was obtained by analyzing the damage threshold circles for various indentation duration from 15 to 300 min. Kruskal–Wallis test, a nonparametric analysis of variance (ANOVA), with 0.05 level of significance was used to test the effects of H2O2 treatment at different indentation durations. The experimental data for myoblasts with or without H2O2 pre-treatment were both fitted with a decreasing Boltzmann-type sigmoid function:17
where K, \(t_{0}\), a and C are empirically determined constants by curve fitting; and \(\sigma (t)\) is the compressive stress threshold. The least square method was used in the curve fitting.
Results
Hyper-visoelastic Material Property
The hyperelastic property of the agarose gel was determined under uniaxial unconfined compression. The result was shown in Fig. 2. First-order Ogden model was used to fit the experimental data. The fitting result in Eq. (1) was as follow: \(\mu_{1} = 1.0142E - 4\), \(\alpha_{1} = 1.76\), \(D_{1} = 3159\). The mean Poisson’s ratio measured at ten equilibrium strains was 0.28.

Equilibrium stress–strain relationship of the agarose gel in uniaxial unconfined compression
The viscoelastic property of the agarose gel was ascertained in the ramp-relaxation experiment under uniaxial unconfined compression. The relaxation curve was fitted with a one-term Prony series to obtain the material constants in Eq. (3): \(\bar{g}_{1}^{\text{P}} = 0.996\), \(\tau_{1}^{\text{G}} = 48.5\).
The experimental measurement of the indentation reaction force and the FEM prediction were shown in Fig. 3 for the validation of the FEM indentation model. While the average difference between the FEM predications and the experimental measurements was about 27%, the two agree extremely well in the response after 10 min.

The experimental indentation force and the FEM simulated data as function of time. Average difference of FEM simulated data from experimental data was about 27%
Indentation Simulation
The radial equilibrium stress field profile at the bottom of the gel obtained from the ABAQUS finite element analysis was shown in Fig. 4. It was noted that the compressive stress at 13.4 mm from the center was almost zero. The stress at distance larger than 13.4 mm became slightly tensile due to the adhered condition assumed in the analysis. Those tensile stresses were three orders of magnitude below the magnitude of compressive stresses. The compressive true strains in the present FEM simulation were roughly about 25% higher than the analytical results reported by Gefen and coworkers for collagen Matrigel based on a transversely isotropic elastic layer model.15

The radial profile of the equilibrium stress field at the bottom of the agarose gel
Damage Stress Threshold
The percentages of cell damage were obtained for three groups of myoblasts monolayers: (I) not covered by agarose gel, (II) covered by agarose gel but without indentation, and (III) covered by agarose gel with 50% indentation for 140 min at the center of the gel. PI staining showed that percentages of cell damage in group I and II were similar at about 3%, showing that the presence of agarose gel had little effect on the percentage of cell damage in the time period studied. Group III showed a maximum of 25.3% cell damage at the center area of the culture dish where stress was highest at about 398 Pa. The percentage of cell damage at the center of the field in Group III was about 8 times of those in Group I and II. The percentage of cell damage decreased radially to about 5.1% at the periphery. It is clear that a 140 min exposure of such indentation could cause a marked increase in the percentage of cell damage beneath the gel under the indenter.
Studies on each group were repeated with n = 3. Sample size of three satisfied the statistical power of 0.8 while controlling type I error at 0.05 for one-tailed test at most time points. The spatial profile of the percentage cell damage of non-treated, 1-h H2O2 and 20-h H2O2 pretreated cells were shown in Fig. 5. With the increase of the indentation duration, the extent of cell damage expanded. Near the center of the indented area, the percentage of cell damage of differently treated cells increased with the increase of the loading duration (Table 2).

The percentage of cell damage of untreated and pre-treated myoblasts derived from the PI fluorescent images taken at different indentation duration
Before indentation, the percentage cell damage for the non-treated myoblasts, and the 0.1 and 0.5 mM H2O2 pre-treated myoblasts for 1 h were all around 3%. The corresponding results were about 5 and 16.2% respectively for the 0.1 and 0.5 mM H2O2 pre-treated myoblasts for 20 h. Thus chronic exposure to higher oxidative stress caused more cell damage without indentation, while short exposure to lower oxidative stress made little difference in cell damage.
The relationship between the compressive stress threshold and the tolerance duration of H2O2-treated and non-treated cells were shown in Figs. 6 and 7. Results indicated that the chronic oxidative stress at higher-dosage could weaken the capability of myoblasts to resist compressive damage (Fig. 6), while short exposure to lower oxidative stress could enhance the capability (Fig. 7). For the indentation durations of 30, 70, and 140 min, Kruskal–Wallis test showed significant differences respectively between the non-treated group and the pre-treated group with 0.5 mM 20-h H2O2 (p < 0.05) (Fig. 6). For the indentation durations of 70, 160, and 180 min, Kruskal–Wallis test showed significant differences between the non-treated group and the pre-treated group with 0.1 mM 1-h H2O2 (p < 0.05). The model parameters obtained from curve-fitting the results of the untreated and the pre-treated groups were shown in Table 3.

The relationships between the compressive stress thresholds and the tolerance durations of myoblasts with 20 h of 0.1 and 0.5 mM H2O2 pretreatments as compared to the non-treatment group. Curve-fitted parameters could be found in Table 3 except the 0.5 mM 20-h H2O2 group. Double sigmoid curve could better fit the results for the 0.5 mM 20-h H2O2 group (R 2 = 0.9999). Orange solid diamonds showed the results from myoblasts being indented in the continued presence of H2O2 without replacing it with fresh normal culture medium

The relationships between the compressive stress thresholds and the tolerance durations of myoblasts with 1 h of 0.1 and 0.5 mM H2O2 pretreatments as compared to the non-treatment group. Curve-fitted parameters could be found in Table 3
It was noted that the curve-fitting results for the 0.5 mM 20-h H2O2 treated myoblasts could be further improved by using a double sigmoid curve (Fig. 6). Equation (5) described mathematically the relationship between the compressive damage threshold and the tolerance duration of H2O2 pre-treated myoblasts with R 2 very close to 1.
Curve-fitting the H2O2 results led to \(K_{0}\) = 110.8 Pa, \(K_{1}\) = 177.8 Pa, C = 6.9 Pa, \(t_{0}\) = 49.36 min, \(t_{1}\) = 124.1 min, \(\alpha_{0}\) = 2.133 min−1, \(\alpha_{1}\) = 0.26 min−1(R 2 = 0.9999).
Discussion
The percentage of cell damage changed sharply along the radius. This apparently corresponded with the profile of compressive stress acting on the cells induced by the indentation on the agarose gel. Using this approach, we have quantitatively measured the damage threshold of myoblasts under compression in term of a threshold curve of stress intensity and loading duration. This damage threshold at the cellular level would underpin a multiscale understanding of deep tissue injury due to tissue internal stresses.
Cell damage increased with the duration of indentation (Table 2). The reason may be due to the gradual breakdown of the cytoskeletal and membrane structure of the cells. It was observed that the tolerance of myoblasts to compressive stress decreased significantly at around 160 min of loading. Similar trends have been reported in term of strain-duration threshold for tissue-engineered bio-artificial muscles (BAMs),15 and in term of pressure-duration threshold in an animal model.17 The myoblasts apparently could tolerate compressive stress below 9 Pa in our current model for at least 5 h which was similar to the corresponding time point of 4.75 h in the BAMs model reported by Gefen.15 The estimated sigmoid step time (\(t_{0} = 160.3\) min) was about 38% larger than the step time in the BAMs model.15 The significant decrease in stress endurance lasted approximately for 40 min in our experiments as compared to the decrease in strain endurance that lasted for about 110 min in the BAMs model. This difference might be due to a number of factors, including the 2D vs. 3D configuration of the experiments. A 2D model is understandably less representative of the in vivo environment.
Chronic oxidative stress caused more cell damage and also significantly weakened the capability of myoblasts to resist compressive damage. H2O2 could contribute to the depolymerization of actin filaments due to the up-regulation of cofilin2 and the down-regulation of thymosin-beta-4.37 As the actin filaments are major components of the cytoskeletal structure, depolymerization of actin filaments would decrease the integrity of cytoskeleton, thus decrease the capability of the cells to resist compressive damage. Short-term oxidative stress caused little cell damage; and short-term exposure to low oxidative stress like 0.1 mM H2O2 could apparently enhance the capability of myoblasts to resist compressive damage. Some evidences show that sublethal exposure to oxidative stress could results in oxidative stress resistance.20 Different levels of oxidative stress may enhance or weaken cells.
In Fig. 6, the results shown as orange solid diamonds were obtained from myoblasts not only pre-treated with 0.5 mM H2O2, but also with indentation conducted in similar oxidative medium, i.e., without replacing the oxidative medium with fresh normal culture medium as originally described in the methodology. The double sigmoid curve seemed to suggest that in about 1–2 h after removing H2O2 from the culture environment, myoblasts could apparently regain some capability to resist compressive damage, although the extended capability still could not reach their original status when compared to the damage threshold curve obtained for the control untreated cells. Such regain in capacity to resist compressive damage was not apparent in the results when the oxidative medium was not removed, as shown by the orange solid diamonds in Fig. 6. We are planning to follow-up with a formal study to ascertain the recovery of the damage threshold of muscle cells in different environments.
From the examples presented in Fig. 5, 0.5 mM H2O2 pre-treatment for 1 h could bring protection against mechanical damage after 180 min of compression, compared to the control. The same protection could not be claimed for long-term (20 h) exposure to both 0.1 and 0.5 mM H2O2. Actually, such chronic exposures could significantly weaken the cells (Figs. 5 and 6). From Fig. 7, one could readily observe that at the 180 min time point, the 0.5 mM 1-h group and 0.1 mM 1-h group both showed a higher compressive damage threshold when compared to the control. At 300 min, the compressive damage thresholds were quite similar among the two oxidative groups and the control, indicating the earlier protections by the short oxidative exposures failed to protect the myoblasts from long-term compressive damages.
The results implied that chronic exposure to high oxidative stress might compromise the capability of muscle cells to withstand compressive damages, while short exposure to mild oxidative stress might enhance such capability. Thus the exact pattern of in vivo oxidative stress environment might be important for DTI development. There are however few literatures on quantitative oxidative stress measurement in muscle tissues in vivo during reperfusion and chronic inflammation. Therefore, accurate in vivo measurement of the intensity and duration of oxidative exposure would help uncover the significances of oxidative stress on the clinical development of DTI.
The limitations of the present study include the following. A 2D monolayer of myoblasts in vitro is certainly different from a 3D muscle tissues in vivo. Cell morphology is highly coupled with cell–environment interactions. Future studies should include the use of 3D models to mimic more closely to the in vivo situations. Primary cells from living muscle tissues may respond quite differently to oxidative and mechanical challenges as compared to C2C12 genetically modified myoblasts. Pilot studies should be conducted in the future to ascertain whether the conclusions made on the C2C12 myoblasts can indeed be confirmed on the primary cells culture. In this paper, we have studied only two exposure durations (1 and 20 h) and two H2O2 concentrations (0.1 and 0.5 mM). More exposure durations and oxidative stress intensities, particularly those obtained in the context of DTI, would need to be studied further in the future.
References
Allman, R. M., P. S. Goode, N. Burst, A. A. Bartolucci, and D. R. Thomas. Pressure ulcers, hospital complications, and disease severity: impact on hospital costs and length of stay. Adv. Skin Wound Care 12(1):22–30, 1999.
Allman, R. M., P. S. Goode, M. M. Patrick, N. Burst, and A. A. Bartolucci. Pressure ulcer risk factors among hospitalized patients with activity limitation. Jama 273(11):865–870, 1995.
Anderson, J. E. A role for nitric oxide in muscle repair: nitric oxide–mediated activation of muscle satellite cells. Mol. Biol. Cell. 11(5):1859–1874, 2000.
Bader, D. L., C. Bouten, D. Colin, and C. W. J. Oomens. Pressure Ulcer Research: Current and Future Perspectives. New York: Springer, 2005.
Bogie, K. M., I. Nuseibeh, and D. L. Bader. Early progressive changes in tissue viability in the seated spinal cord injured subject. Spinal Cord 33(3):141–147, 1995.
Breuls, R. G. M., C. V. C. Bouten, C. W. J. Oomens, D. L. Bader, and F. P. T. Baaijens. Compression induced cell damage in engineered muscle tissue: an in vitro model to study pressure ulcer aetiology. Ann. Biomed. Eng. 31(11):1357–1364, 2003.
Breuls, R. G. M., A. Mol, R. Petterson, C. W. J. Oomens, F. P. Baaijens, and C. V. Bouten. Monitoring local cell viability in engineered tissues: a fast, quantitative, and nondestructive approach. Tissue Eng. 9(2):269–281, 2003.
Byrne, D. W., and C. A. Salzberg. Major risk factors for pressure ulcers in the spinal cord disabled: a literature review. Spinal Cord 34:255–263, 1996.
Choudhury, N. A., S. Sakaguchi, K. Koyano, A. F. M. Matin, and H. Muro. Free radical injury in skeletal muscle ischemia and reperfusion. J. Surg. Res. 51(5):392–398, 1991.
Coleman, B. D., and D. C. Newman. On the rheology of cold drawing. II. Viscoelastic materials. J. Polym. Sci. Part B: Polym. Phys. 30(1):25–47, 1992.
Coleman, S., J. Nixon, J. Keen, L. Wilson, E. McGinnis, C. Dealey, N. Stubbs, A. Farrin, D. Dowding, J. M. G. A. Schols, J. Cuddigan, D. Berlowitz, E. Jude, P. Vowden, L. Schoonhoven, D. L. Bader, A. Gefen, C. W. J. Oomens, and E. A. Nelson. A new pressure ulcer conceptual framework. J. Adv. Nurs. 70(10):2222–2234, 2014.
Deseri, L., M. Fabrizio, and M. Golden. The concept of a minimal state in viscoelasticity: new free energies and applications to PDEs. Arch. Ration. Mech. Anal. 181(1):43–96, 2006.
Federico, A., F. Morgillo, C. Tuccillo, F. Ciardiello, and C. Loguercio. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer. 121(11):2381–2386, 2007.
Gawlitta, D., W. Li, C. W. J. Oomens, F. P. Baaijens, D. L. Bader, and C. V. Bouten. The relative contributions of compression and hypoxia to development of muscle tissue damage: an in vitro study. Ann. Biomed. Eng. 35(2):273–284, 2007.
Gefen, A., B. van Nierop, D. L. Bader, and C. W. J. Oomens. Strain-time cell-death threshold for skeletal muscle in a tissue-engineered model system for deep tissue injury. J. Biomech. 41(9):2003–2012, 2008.
Gissel, H. The role of Ca2+ in muscle cell damage. Ann. N. Y. Acad. Sci. 1066(1):166–180, 2006.
Linder-Ganz, E., S. Engelberg, M. Scheinowitz, and A. Gefen. Pressure–time cell death threshold for albino rat skeletal muscles as related to pressure sore biomechanics. J. Biomech. 39(14):2725–2732, 2006.
Linder-Ganz, E., and A. Gefen. Mechanical compression induced pressure sores in rat hindlimb: muscle stiffness, histology, and computational models. J. Appl. Physiol. 96:2034–2049, 2004.
Linder-Ganz, E., and A. Gefen. Stress analyses coupled with damage laws to determine biomechanical risk factors for deep tissue injury during sitting. J. Biomech. Eng. 131.1:011003, 2009.
Liochev, S. I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 60:1–4, 2013.
Mak, A. F. T., Y. Y. Yu, L. P. C. Kwan, L. Sun, and E. W. C. Tam. Deformation and reperfusion damages and their accumulation in subcutaneous tissues during loading and unloading: a theoretical modeling of deep tissue injuries. J. Theor. Biol. 289:65–73, 2011.
Mak, A. F. T., M. Zhang, and E. W. C. Tam. Biomechanics of pressure ulcer in body tissues interacting with external forces during locomotion. Annu. Rev. Biomed. Eng. 12:29–53, 2010.
McNeil, P. Membrane repair redux: redox of MG53. Nat. Cell. Biol. 11:7–9, 2009.
Ogden, R. W. Large deformation isotropic elasticity-on the correlation of theory and experiment for incompressible rubberlike solids. Proc. R. Soc. Lond. A. 326(1567):565–584, 1972.
Oomens, C. W. J., S. Loerakker, and D. L. Bader. The importance of internal strain as opposed to interface pressure in the prevention of pressure related deep tissue injury. J. Tissue Viability 19(2):35–42, 2010.
Peeters, E. A. G., C. W. J. Oomens, C. V. C. Bouten, D. L. Bader, and F. P. T. Baaijens. Mechanical and failure properties of single attached cells under compression. J. Biomech. 38(8):1685–1693, 2005.
Peirce, S. M., T. C. Skalak, and G. T. Rodeheaver. Ischemia-reperfusion injury in chronic pressure ulcer formation: a skin model in the rat. Wound Repair Regen. 8(1):68–76, 2000.
Pullarkat, P. A., P. A. Fernández, and A. Ott. Rheological properties of the eukaryotic cell cytoskeleton. Phys. Rep. 449(1):29–53, 2007.
Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging. 2(2):219, 2007.
Siu, P. M., E. W. Tam, B.T. Teng, X.M. Pei, J.W. Ng, I.F. Benzi, and A.F. Mak. Muscle apoptosis is induced in pressure-induced deep tissue injury. J. Appl. Physiol. 107:1266–1275, 2009.
Siu, P. M., Y. Wang, and S. E. Alway. Apoptotic signaling induced by H2O2-mediated oxidative stress in differentiated C2C12 myotubes. Life Sci. 84(13):468–481, 2009.
Slomka, N., S. Or-Tzadikario, D. Sassun, and A. Gefen. Membrane-stretch-induced cell death in deep tissue injury: computer model studies. Cell Mol. Bioeng. 2(1):118–132, 2009.
Solis, L. R., A. Liggins, R. R. Uwiera, N. Poppe, E. Pehowich, P. Seres, R. B. Thompson, and V. K. Mushahwar. Distribution of internal pressure around bony prominences: implications to deep tissue injury and effectiveness of intermittent electrical stimulation. Ann. Biomed. Eng. 40(8):1740–1759, 2012.
Stroetz, R. W., N. E. Vlahakis, B. J. Walters, M. A. Schroeder, and R. D. Hubmayr. Validation of a new live cell strain system: characterization of plasma membrane stress failure. J. Appl. Physiol. 90(6):2361–2370, 2001.
Sun, S., S. Wong, A. Mak, and M. Cho. Impact of oxidative stress on cellular biomechanics and the rho signaling in C2C12 myoblasts. J. Biomech. 47(15):3650–3656, 2014.
Warren, G. L., M. Summan, X. Gao, R. Chapman, T. Hulderman, and P. P. Simeonova. Mechanisms of skeletal muscle injury and repair revealed by gene expression studies in mouse models. J. Physiol. 582(2):825–841, 2007.
Wong, S. W., S. Sun, M. Cho, K. K. Lee, A. F. T. Mak. H2O2 exposure affects myotube stiffness and actin filament polymerization. Ann. Biomed. Eng. 1–11, 2014.
Wozniak, M. A., and C. S. Chen. Mechanotransduction in development: a growing role for contractility. Nat. Rev. Mol. Cell Biol. 10(1):34–43, 2009.
Xiao, D. Z. T., S. Y. Q. Wu, and A. F. T. Mak. Accumulation of loading damage and unloading reperfusion injury—modeling of the propagation of deep tissue ulcers. J. Biomech. 47(7):1658–1664, 2014.
Acknowledgments
This study was supported by Hong Kong Research Grant Council (RGC Ref. No.: CUHK415413).
Conflict of interest
The authors of the above paper state that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Amit Gefen oversaw the review of this article.
Rights and permissions
About this article

Cite this article
Yao, Y., Xiao, Z., Wong, S. et al. The Effects of Oxidative Stress on the Compressive Damage Thresholds of C2C12 Mouse Myoblasts: Implications for Deep Tissue Injury. Ann Biomed Eng 43, 287–296 (2015). https://doi.org/10.1007/s10439-014-1239-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-014-1239-6