
Abstract
The ultraviolet (UV) radiation from sunlight exposure is highly related to skin damages such as photoaging and skin cancers. UVA radiation can cause altered expression of extracellular matrix proteins, and UVB is believed to be an apoptosis induction factor. In the past, many experiments have been conducted in vitro to investigate how UV lights are responsible for cell damages. However, most of these studies were done under static conditions such as culture dishes, which were quite different from the in vivo dynamic circulatory system. Also, in these studies, only one UV dose was applied at one time, limiting the experimental throughput. To best mimic the physiological condition as well as to increase the throughput, a microfluidic chip was designed and fabricated to create five different UV doses in one single experimental run. This biocompatible chip is used for seeding and culturing cells, together with observing cell morphology in a circulating condition. By using the “Christmas tree” structure, five different concentrations of blue dye solutions were generated within a microfluidic chip. And by illuminating this chip with the UV light, five different UV doses could be attained on cells cultured within this chip. The effects of UVB dose on NIH/3T3 fibroblasts, mainly in terms of morphology and fluorescence-based cell damage rate, were studied.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The UV radiation from sunlight exposure has both positive and negative effects on human health. Adequate amounts of sunlight ensure normal production of Vitamin D by the body, which is essential in strengthening bones (Cranney et al. 2008; Hacker et al. 2011; Morris et al. 2010; Reid et al. 2014) and possibly inhibiting the growth of some cancers (Chakraborti 2011; Picotto et al. 2012). Also, UV exposure increases the amount of melanin in the skin. This brown pigment has an excellent photoprotective effect by absorbing UV radiation, dissipating the energy, and protecting the skin from DNA damages (Kvam and Dahle 2004; Miyamura et al. 2008; Wakamatsu and Ito 2006). On the other hand, excessive sun exposure is known to be associated with photoaging, skin cancers, immune suppression, and eye diseases such as cataracts. The World Health Organization (WHO) has classified the broad-spectrum UV radiation as a Group 1 carcinogen.
Depending on the wavelength, UV lights can be divided into UVA (315–400 nm), UVB (280–315 nm), and UVC (100–280 nm). While UVA, also referred to as the black light, cannot be absorbed by the ozone layer, UVB and UVC can be mostly and completely absorbed by the ozone shield, respectively. UVA has long been believed to play an important role in skin aging (Boisnic et al. 2005; Haywood et al. 2003; Kim et al. 2013; Krutmann 2003; Yin et al. 2001). Battie et al. (2014) reported that UVA radiation is a key role in pigmented changes occurring with age, the major sign of skin photoaging in Asians.
Although UVB phototherapy is a common modality for psoriasis and other skin diseases, overexposure of UVB can cause direct DNA damage, leading to some forms of skin cancers such as basal cell carcinoma (BCC), squamous cell carcinoma (SCC), and melanoma (Green et al. 2007; Hearn et al. 2008; Lee et al. 2005; Saraiya et al. 2004). Situm et al. (2008) reported that a latency period of 20–50 years is typical between the time of UV damage and the clinical onset of BCC. Also, UV-induced mutations in the TP53 tumor suppressor gene have been found in about 50% of BCC patients. Jhappan et al. (2003) reviewed the correlation between UV radiation and cutaneous malignant melanoma and concluded that in vivo models in mice and human skin xenografts have contributed significantly toward the development of effective approaches to the prevention and treatment of melanoma.
Considering the effects of UVC, the eye is most sensitive to this wave band, and overexposure to this radiation can result in eye damages such as cataracts, pterygium, and pinguecula. By examining more than 100,000 Aborigines and non-Aborigines in rural Australia, a strong positive correlation between climatic UV radiation and pterygium prevalence was found (Moran and Hollows 1984). The impacts of UV radiation on eye diseases are reviewed in references (Behar-Cohen et al. 2014; Bergmanson and Soderberg 1995; Parisi et al. 2001).
To help people effectively protect themselves from UV radiation, an international standard UV index is designed to measure the strength of UV lights from sunlight exposure. This index is directly proportional to UV intensity, and it can be classified as follows: 0–2 (low, intensity = 0–20 W/m2); 3–5 (moderate, intensity = 21–35 W/m2); 6–7 (high, intensity = 36–45 W/m2); 8–10 (very high, intensity = 46–55 W/m2); and ≧11 (extreme, intensity ≧ 56 W/m2). Therefore, to quantitatively study the effects of UV radiation on cells, controllable UV intensity (in W/m2) and dose (in J/m2) are in demand.
Traditionally, experiments showing how UV lights could cause cell damages were conducted in static, one-dose only conditions. This means that cells were cultured in petri dishes, microplates or glasses, and subjected to one UV dose at one time. For example, Kimura et al. (2010) investigated the cell-killing efficacy of UV light on fluorescent protein-expressing cancer cells in vitro and in vivo. Four types of cancer cells (human fibrosarcoma, human osteosarcoma, lung carcinoma, and human pancreas cancer) were cultured on cover glasses and irradiated with UV lights from the bottom of the substrate. It was shown that (1) most cells were viable after an UVA exposure of 200 J/m2, (2) cell death was observed with UVB irradiation at 50 J/m2, and (3) 25 J/m2 UVC irradiation killed almost 70% of the human osteosarcoma cells. Using NIH/3T3 fibroblasts and A431 epidermoid carcinoma cells grown in 12-well microplates as models, Tai et al. (2012) reported that UVC is a DNA damage inducer, and 20 J/m2 of UVC irradiation caused cell growth inhibition as well as induced cell death after 24–36-h exposure. Bryant et al. (2000) studied the cytocompatibility of UV light photoinitiating system on NIH/3T3 fibroblasts cultured in 12- and 24-well microplates.
All above-mentioned and mostly other studies were performed in static conditions where cells didn’t subject to fluidic flowing like they do in the in vivo microenvironment. Lo et al. (2013) found that lung cancer cells responded to an antioxidant, α-tocopherol, differently depending on circulating or static conditions they were cultured in. Also, in these studies, cells were irradiated with one UV intensity/dose at one time, limiting the throughput of the experiment.
In order to systematically and quantitatively study how NIH/3T3 fibroblasts respond to UV lights, a microfluidic chip was designed and fabricated to mimic the physiologically circulating condition as well as to increase the experimental throughput. This chip is biocompatible for seeding and culturing cells, and it is transparent for observation of cell morphology and staining. Applying a “Christmas tree”-shaped structure to mix and redistribute solutions, five relative concentrations of 0, 1/8, 1/2, 7/8, and 1 are achieved above the culture regions (Lo et al. 2013). A blue colorant, Brilliant Blue FCF, was used to shield off different amounts of UV lights in the five culture areas. The shielding ratio and therefore cell-exposed UV intensity can be well controlled by adjusting the concentration of the blue dye from the inlet. With the aid of this microfluidic chip, we were able to study the effects of UV dose on NIH/3T3 fibroblasts in a circulating condition and a high-throughput manner.
2 Materials and methods
2.1 Chip design and fabrication
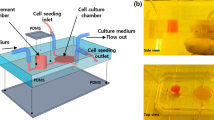
The design of the microfluidic chip is shown in Fig. 1. The pattern was drawn in AutoCAD (Autodesk) and then loaded into a CO2 laser scriber (ILS2, Laser Tools & Technics Corp.) to ablate desired patterns on polymethylmethacrylate (PMMA) substrates and double-sided tapes (8018, 3 M). As shown in Fig. 1a, five layers of PMMA substrates (thickness = 1 mm) and double-sided tapes (thickness = 260 and 70 μm) were bound together to form the integrated chip, which was then attached to a petri dish (Nunc) for cell culture. The top PMMA layer had five small holes/adaptors serving as flow inlets and outlets. The next two double-sided tapes provided fluidic channels for the shielding area, having a width of 1 mm and a total thickness of 330 μm (Fig. 1b left). The fourth PMMA layer connected the culture area with the topmost flow inlet/outlet. The bottom double-sided tape (thickness = 260 μm) served as the culture area (Fig. 1b right). Cytotoxicity of PMMA substrates on cells was examined, and no significant change in cell viability was observed (Huang et al. 2009).

Design of the microfluidic chip. a From left to right: 1-mm PMMA layer with adaptors, 260-μm double-sided tape (shielding area), 70-μm double-sided tape (shielding area), 1-mm PMMA layer, 260-μm double-sided tape (culture area), and petri dish (culture area). b Fluidic channels of the shielding area (left) and the culture area (right)
2.2 Calculation and simulation of concentrations and absorbance
As shown in Fig. 1b right, when two solutions with relative concentrations of 1 and 0 were injected from the two dye/water inlets, relative concentrations of 0, 0.125, 0.5, 0.875, and 1 could be achieved in the five rectangular areas near the waste outlet. Here, we assume that all liquids split-flowed smoothly and equally around the fork. Similar “Christmas tree” structures were reported for evaluating toxicity and teratogenicity of drugs on embryonic zebrafish developmental dynamics (Yang et al. 2011), for studying the effects of antioxidant concentrations on the production of reactive oxygen species (ROS) in lung cancer cells (Lo et al. 2013), and for investigating the migration of lung cancer cells under single or coexisting chemical/electrical stimulation (Lo et al. 2016). In the present study, 0.5% (w/w in water) Brilliant Blue FCF (Sigma) was used as the UV blocker, creating concentrations of 0, 0.0625, 0.25, 0.4375, and 0.5% above five locations of the culture area. In addition, the numerical simulation of chemical concentrations inside the microfluidic chip was performed using the commercial software COMSOL Multiphysics (COMSOL, USA). The “Transport of Dilute Species” module was used with the following parameters and settings: medium = water; diffusion coefficient = 10−13 m2/s for Brilliant Blue FCF at room temperature (Gupta et al. 2006); inlet flow rate = 15 μL/h (set in each inlet).
The Beer’s law was used to relate the attenuation of light to the properties of the material through which the light is traveling. The absorbance (A) of a material to a specific light band is proportional to the absorption coefficient of the material (α), the path length of the light (x), and the concentration of the material (c): A = αxc. Also, the absorbance is related to the transmittance (T) of the light to the material as A = −logT. Using these two equations, we can relate the absorbance (A) to the concentration (c) by measuring the light intensity underneath the shielding area and normalizing the transmittance to 1 for 0% Brilliant Blue FCF (i.e., the light was totally transmitted through the area without any UV blocker).
2.3 Cell preparation
The fibroblast cell line NIH/3T3 was purchased from the Bioresource Collection and Research Center (BCRC), Taiwan. A complete medium consisting of Dulbecco’s modified Eagle medium (DMEM, Gibco) and 10% calf serum (CS, Invitrogen) was used for the cell culture. The cells were incubated in tissue culture polystyrene flasks (corning) in 5% CO2 at 37 °C until 90% confluence before seeding into the microfluidic chip.
2.4 Experimental system and procedure
2.4.1 Chip assembly and cell preparation
The microfluidic chip was assembled inside the laminar flow hood. The medium/cell inlet and outlet were connected to two syringes driven by syringe pumps (NE-300, New Era). 1 × phosphate-buffered saline (PBS) was flowed into the chip and kept within the channel overnight under 5% CO2 at 37 °C. Then, the syringes were pushed gently by hands to remove bubbles, and 1 × PBS was replaced with DMEM. 4 × 105 cells suspended in 1 ml DMEM with 10% CS was loaded into the culture area by injecting the solution from the medium/cell inlet. The microfluidic chip with seeded cells was mounted on top of a transparent indium tin oxide (ITO) glass (Part No. 300739, Merck) which is connected to a proportional–integral–derivative (PID) controller (TTM-J4-R-AB, JETEC Electronics Co.) for maintaining the temperature at 37 ± 0.5 °C via feedback from a thermal couple (TPK-02A, TECPEL) clamped tightly between the heater and the chip. Fresh medium (DMEM with 10% CS) was continuously pumped into the chip at a flow rate of 20 μL/h for 24 h before UVB treatments. The continuous flowing of fresh medium provides cells with enough nutrition as well as removes waste from cells. In vivo cells are subject to different flow rates in different body fluids such as urine, blood, saliva, and tears. In the present study, the flow rate is determined to be 20 μL/h based on our previous experience (Cheng et al. 2008; Lo et al. 2013; Sun et al. 2012; Wu et al. 2015). This rate should not be too high to affect the metabolism of cells. As reported, fluidic shear stresses of as low as 0.25–0.6 Pa could interface cell attachment, and even higher values of stresses (0.5–10 Pa) could remove adherent cells (Chisti 2001). Moreover, 0.8–1.5 Pa of laminar shear stresses could induce cell alignment in the direction of flow (Davies et al. 1986), and values of 0.1–1 Pa were shown to affect cellular morphology and permeability (Chisti 2001). A flow rate of 20 μL/h corresponds to a shear stress of 0.0001 Pa (Sun 2016), which is considered harmless to cells. Also, this flow rate is high enough for providing cells with fresh medium since cells remain healthy inside the culture area for days (Cheng et al. 2008).
2.4.2 UV treatments
As indicated in our previous studies (Wu et al. 2016), UVC, compared to UVB, had a much lower transmittance to plastic materials such as petri dishes and microplates. Therefore, UVB was used as the UV light source in the present work. Before UV treatments, two syringes containing Brilliant Blue FCF of 0 and 0.5% were connected to the water/dye inlets of the microfluidic chip and driven by pumps at a flow rate of 15 μL/min (set in each inlet) for at least 30 min. Water and colorant solutions were kept flowing into and out of the chip during UV treatments to ensure uniform Brilliant Blue FCF concentrations in the shielding area. The UV lamp (3UV Lamps, UVP LLC) was mounted 3 cm above the chip and turned on for 22 min and 44 s, yielding UVB doses of 0.604, 0.423, 0.091, 0.035, and 0 J/cm2 underneath the five shielding areas. These doses were calculated by multiplying the power (in W/cm2), measured by a UV-specified power meter (UVX Radiometer, UVP LLC), by the total illumination time (in sec). Bright-field images of different field of views (FOVs) were captured using an inverted microscope (CKX41, Olympus) before UV treatments, right after UV treatments, 1, 2, 4, 10, and 24 h after UV treatments.
2.4.3 Cell staining
To quantify the effect of UVB dose on cell damage, cell staining with Hoechst and SYTOX Green was performed. Hoechst (H3570, Thermo), having excitation and emission wavelengths of around 350 and 461 nm, respectively, was used to stain double-stained DNAs in all cells. SYTOX Green (S7020, Thermo), having excitation and emission wavelengths of around 504 and 523 nm, respectively, was used to stain nucleic acids in damaged cells. 24 h after UV treatments, cultured cells inside the microfluidic chip were incubated with Hoechst (8 μM in DMEM) and SYTOX Green (0.2 μM in DMEM) for 20 min and then washed with fresh DMEM 3 times. Fluorescent images of different FOVs were captured using an inverted fluorescent microscope (TS-100, Nikon) equipped with a CCD camera (DSQi1, Nikon).
2.5 Data analysis
For each experimental condition, at least 120 cells were selected from at least three independent experiments for data analysis. The standard error of the mean (SEM) was also calculated. To quantify cell morphology, each cell was assumed to be elliptic, and its long axis (L 1) and short axis (L 2) were measured by ImageJ software (National Institute of Health), as shown in Fig. 2. Then, the long axis-to-short axis ratio, L 1/L 2, was calculated. To analyze cell-staining images, the numbers of all cells (those stained with Hoechst) and damaged cells (those stained with SYTOX Green) were counted in ImageJ software. The damage rate is defined as (the number of damaged cells)/(the number of all cells) × 100%.

Long axes (L 1) and short axes (L 2) of NIH/3T3 cells before (left) and after (right) UVB exposure
3 Results and discussion
3.1 Calculation and simulation of concentrations and absorbance
Figure 3a shows the model used in the simulation with the mesh distribution shown in Fig. 3b. There are 39,052 prism elements, 25,712 tetrahedral elements, 19,960 triangular elements, 4872 edge elements, 774 vertex elements, 432 pyramid elements, and 84 quadrilateral elements. Figure 3c shows the numerical simulation of Brilliant Blue FCF concentration inside the microfluidic chip, where the concentrations in the two inlets are 0 and 0.5%, respectively. As indicated, the concentrations are close to calculated values, being 0, 0.0625, 0.25, 0.4375, and 0.5%. To experimentally verify the distribution of concentration, 0 and 0.5% Brilliant Blue FCF were continuously flowed into the two inlets of the chip for 1 h, and the whole chip was imaged using an inverted microscope, as shown in Fig. 3d. The absolute intensity in each shielding area was measured using ImageJ, and these values (the y-axis) were plotted against the calculated ones (the x-axis). A good linear correlation was obtained between calculation and measurement (R 2 = 0.9962) (see Fig. 3e).

a Model used in the simulation. b Mesh distribution in the simulation. c Numerical simulation of Brilliant Blue FCF concentration inside the microfluidic chip. The concentrations in the two inlets are 0.5 and 0%, respectively. d Image of the microfluidic chip with 0.5 and 0% of Brilliant Blue FCF solutions continuously flowing into the two inlets. e Measured intensities plotted against calculated concentrations
The UVB intensity underneath each shielding area was measured as the light source passed through and blocked by the colorant. As shown in Fig. 4a, these values were around 443 ± 5.5, 310 ± 4.4, 67 ± 3.6, 26 ± 0.0, and 23 ± 0.0 μW/cm2 for colorant concentrations of 0, 0.0625, 0.25, 0.4375, and 0.5%, respectively (averaged over three measurements). After normalizing the transmittance to 1 for 0% Brilliant Blue FCF, the absorbance was calculated (Table 1) and plotted against the concentration, as shown in Fig. 4b. For example, for concentration (c) = 0.0625%, the normalized transmittance (T) was 310.3908/443.0049 = 0.7006487, and the absorbance (A) was −log0.7006487 = 0.1545. A good linear correlation of R 2 = 0.9905 indicated that within this concentration range Beer’s law is valid and absorbance of other values can be achieved using interpolation.

a UVB power densities measured underneath five shielding areas. b Relationship between Brilliant Blue FCF concentration and UVB absorbance
3.2 Effects of UVB on cell morphology
After UVB treatments for 22 min and 44 s, cells cultured underneath five shielding areas were subject to UVB doses of 0.604, 0.423, 0.091, 0.035, and 0 J/cm2, respectively. To quantify cell morphology, the long axis-to-short axis ratios of cells subject to different UVB doses at different time points were measured and calculated. These ratios served as an indicator to determine whether these cells were healthy or not: healthy NIH/3T3 cells appear spindle-like with higher L 1/L 2 ratios, while damaged NIH/3T3 cells look round with lower L 1/L 2 ratios (L 1/L 2 = 1 for a circle) (Coleman et al. 2000; Stacey et al. 1999; Sugita et al. 1992). For example, Coleman et al. reported that bFGF (a fibroblast growth factor)-treated NIH/3T3 cells tended to be smaller and rounder than the typical flat morphology of NIH/3T3 cells (Coleman et al. 2000). Other types of cells also appear round or oval during apoptosis (Elmore 2007; Imajoh et al. 2004; Miller et al. 1998). Therefore, the long axis-to-short axis ratio can be used to quantify the extent to which NIH/3T3 cells were damaged due to UVB treatments. The aspect ratio values for “before UV treatment” cells were 3.968, 4.574, 4.079, and 4.021 for UVB doses of 0.604, 0.423, 0.091, and 0.035 J/cm2, respectively. Clearly, cells show a certain degree of deviation in the aspect ratio in these control cases. Such deviation is considered acceptable since cells cultured in different areas show certain randomness. We assume that cells start to become unhealthy/damaged once the aspect ratio is smaller than around four.
Figure 5a shows these ratios for cells subject to 0.604 J/cm2 UVB radiation at different time points. The mean ratios were 3.968, 3.479, 2.948, 2.918, 2.913, 2.841, and 2.691 before UV treatment, right after UV treatment, 1, 2, 4, 10, and 24 h after UV treatments, respectively. Clearly, this ratio decreased after cells were subject to UVB radiation, and the value decreased even more with increasing post-treatment time. This result suggests that UVB of a 0.604 J/cm2 dose did cause damage to NIH/3T3 cells, more or less in a post-treatment time-dependent manner. The longer the post-treatment time was, the more the cells were damaged (shown as a decreasing long axis-to-short axis ratio). A similar trend was observed for cells subject to 0.423 J/cm2 UVB radiation, as indicated in Fig. 5b. The mean ratios were 4.574, 3.996, 3.737, 3.792, 3.52, 3.357, and 2.729 before UV treatment, right after UV treatment, 1, 2, 4, 10, and 24 h after UV treatments, respectively. Roughly speaking, this ratio decreased after cells were subject to UVB radiation, and the value decreased even more with increasing post-treatment time except at time points 1 and 2 h. For cells subject to UVB doses of 0.091 J/cm2 or less, UVB treatments did not do significant damage to them at post-treatment time points 0, 1, 2, and 4 h. At time points 10 and 24 h, the long axis-to-short axis ratio started to decrease, indicating clear damage to cells. For example, under a UVB dose of 0.091 J/cm2, the mean ratios were 4.079, 4.312, 4.178, 4.288, 4.342, 3.909, and 3.271 before UV treatment, right after UV treatment, 1, 2, 4, 10, and 24 h after UV treatments, respectively. And under a UVB dose of 0.035 J/cm2, the mean ratios were 4.021, 4.469, 4.307, 4.461, 4.256, 4.191, and 3.495 before UV treatment, right after UV treatment, 1, 2, 4, 10, and 24 h after UV treatments, respectively. These results indicate that the onset time point for UVB light (with a dose less than 0.091 J/cm2) to do significant damage to NIH/3T3 cells is around 10 h after treatments. As a control group, under a UVB dose of 0 J/cm2, the mean ratios were 4.387, 4.624, 4.412, 4.608, 4.750, 4.508, and 4.291 before UV treatment, right after UV treatment, 1, 2, 4, 10, and 24 h after UV treatments, respectively. Without UVB exposure, no clear damage to cells was observed.

Long axis-to-short axis ratios of NIH/3T3 cells under a 0.604 J/cm2 and b 0.423 J/cm2 UVB exposure at different time points
Figure 6a shows the long axis-to-short axis ratios of NIH/3T3 cells 10 h after UVB treatments at different doses. This ratio was close to 4.2 before UV exposure, but decreased to 4.191, 3.909, 3.357, and 2.841 after subject to UVB doses of 0.035, 0.091, 0.423, and 0.604 J/cm2, respectively. As shown in Fig. 6b, a similar trend was observed for cells 24 h after UVB treatments at different doses. These ratios decreased to 3.495, 3.271, 2.729, and 2.691 after subject to UVB doses of 0.035, 0.091, 0.423, and 0.604 J/cm2, respectively. These suggest that, after the onset time point of 10 h post-treatment, UVB did damage NIH/3T3 cells in a dose-dependent manner.

Long axis-to-short axis ratios of NIH/3T3 cells a 10 h and b 24 h after UVB exposure at different doses
3.3 Effects of UVB on cell damage rate
Cell damage rates due to UVB treatments were measured and calculated by staining cells with Hoechst and SYTOX Green. Hoechst dyes are permeable to cell membrane and are commonly used to stain double-strained DNAs in cells. SYTOX Green dyes, on the contrary, are permeable only to damaged membrane, and are applied to stain nucleic acids in damaged cells. Figure 7 shows the images of NIH/3T3 cells 24 h after UVB exposure at different doses. Cells in the bright-field images (Fig. 7 left column) can be mapped to those in the Hoechst images (Fig. 7 middle column, expressed in blue), and damaged cells were indicated in the SYTOX Green images (Fig. 7 right column, expressed in green). These images were further quantified by calculating the damaging rate, defined as the number of cells stained with SYTOX Green divided by the number of all cells (stained with Hoechst) in percentage. Figure 8a shows the damage rates 24 h after UVB treatments at different doses. These values were 3.1, 3.6, 3.9, 41.6, and 64.4% for UVB doses of 0, 0.035, 0.091, 0.423, and 0.604 J/cm2, respectively. The damage rate increased with increasing UVB dose from 0.423 to 0.604 J/cm2, and this value stayed almost constant for UVB doses below 0.091 J/cm2.

Images of NIH/3T3 cells 24 h after UVB exposure at different doses. Left column Bright-field; Middle column Hoechst staining; Right column SYTOX Green staining

a Damage rates of NIH/3T3 cells 24 h after UVB exposure at different doses. b Correlation between the long axis-to-short axis ratio and the damage rate
Figure 8b illustrates the correlation between the long axis-to-short axis ratio and the damage rate. As indicated, no linear correlation was observed, and significant damage rates (higher than 40%) were observed for ratios lower than 3. When subject to UVB radiation, cell morphology changed gradually from spindle-like (higher long axis-to-short axis ratios) to round (lower long axis-to-short axis ratios) in both dose- and post-treatment time-dependent manners. But in calculating the damage rate, cells were counted as either damaged or not by determining whether they lighted up or not after SYTOX Green staining. Therefore, the long axis-to-short axis ratio could be used to indicate the extent to which cells were damaged, yet the damage rate showed only the percentage of damaged cells. These results suggest that the threshold of the long axis-to-short axis ratio to initiate observable SYTOX Green signals is around 3. UVB radiation was doing damage to cells gradually with ratios decreasing from 4.5 to 3, but these cells showed “not damaged” in staining. As the dose- or the post-treatment time increased, cells were damaged ever more with ratios further decreasing below 3, while these cells showed “damaged” in staining.
4 Conclusion
In this study, we developed a microfluidic chip for studying the effects of UVB radiation on NIH/3T3 cells. Five different concentrations of Brilliant Blue FCF were produced inside a single chip. Cells were subject to UVB treatments of different doses after the light source passed through the shielding area. By analyzing the cell morphology in terms of the long axis-to-short axis ratio, it was found that UVB radiation did damage to cells in both dose- and post-treatment time-dependent manners. And from cell staining, the damage rates were calculated at different UVB doses, indicating significant damage at UVB doses higher than 0.423 J/cm2. To generate lights with different intensities, a graduated neutral density filter or a spatial light modulator can be used. Although these devices are commercially available and easily to be operated, the present microfluidic chip has the following advantages: (1) five different UV intensities could be attained simultaneously by using this device, highly increasing the experimental throughput, (2) by varying the concentration of the dye within this device, different UV intensities of a linear range could be attained following the calibration curve, and (3) it integrates a microenvironment where the intensity of the UV light is well controlled into a fluidic system where cells are subject to a physiologically circulating condition. In the future, by combining the current chip with other microfluidic designs, we are able to study cells’ response to different chemical or physical stimuli (such as chemicals, lights, electric fields, shear stresses, and magnetic fields) in a high-throughput manner. This will definitely help in understanding the mechanisms of all aspects of cell metabolism.
References
Battie C, Jitsukawa S, Bernerd F, Del Bino S, Marionnet C, Verschoore M (2014) New insights in photoaging, UVA induced damage and skin types. Exp Dermatol 23:7–12
Behar-Cohen F et al (2014) Ultraviolet damage to the eye revisited: eye-sun protection factor (E-SPF(R)), a new ultraviolet protection label for eyewear. Clin ophthalmol 8:87–104. doi:10.2147/OPTH.S46189
Bergmanson JP, Soderberg PG (1995) The significance of ultraviolet radiation for eye diseases. A review with comments on the efficacy of UV-blocking contact lenses. Ophthalmic Physiol Opt J Br Coll Ophthalmic Opt 15:83–91
Boisnic S, Branchet-Gumila MC, Nocera T (2005) Comparative study of the anti-aging effect of retinaldehyde alone or associated with pretocopheryl in a surviving human skin model submitted to ultraviolet A and B irradiation. Int J Tissue React 27:91–99
Bryant SJ, Nuttelman CR, Anseth KS (2000) Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH/3T3 fibroblasts in vitro. J Biomat Sci-Polym E 11:439–457
Chakraborti CK (2011) Vitamin D as a promising anticancer agent. Indian J Pharmacol 43:113–120
Cheng JY, Yen MH, Kuo CT, Young TH (2008) A transparent cell-culture microchamber with a variably controlled concentration gradient generator and flow field rectifier. Biomicrofluidics 2:24105. doi:10.1063/1.2952290
Chisti Y (2001) Hydrodynamic damage to animal cells. Crit Rev Biotechnol 21:67–110. doi:10.1080/20013891081692
Coleman AB, Momand J, Kane SE (2000) Basic fibroblast growth factor sensitizes NIH 3T3 cells to apoptosis induced by cisplatin. Mol Pharmacol 57:324–333
Cranney A, Weiler HA, O’Donnell S, Puil L (2008) Summary of evidence-based review on vitamin D efficacy and safety in relation to bone health. Am J Clin Nutr 88:513s–519s
Davies PF, Remuzzi A, Gordon EJ, Dewey CF Jr, Gimbrone MA Jr (1986) Turbulent fluid shear stress induces vascular endothelial cell turnover in vitro. Proc Natl Acad Sci USA 83:2114–2117
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516. doi:10.1080/01926230701320337
Green A et al (2007) The International Agency for Research on Cancer Working Group on artificial ultraviolet (UV) light and skin cancer. The association of use of sunbeds with cutaneous malignant melanoma and other skin cancers: a systematic review (vol 120, pg 1116, 2007). Int J Cancer 120:2526
Gupta VK, Mittal A, Krishnan L, Mittal J (2006) Adsorption treatment and recovery of the hazardous dye, Brilliant Blue FCF, over bottom ash and de-oiled soya. J Colloid Interf Sci 293:16–26
Hacker A, Fung E, Atkinson S, King J (2011) Maternal vitamin D status and its effect on maternal and infant bone health: a systematic review. Faseb J 25, Article No. 996.10
Haywood R, Wardman P, Sanders R, Linge C (2003) Sunscreens inadequately protect against ultraviolet-A-induced free radicals in skin: implications for skin aging and melanoma? J Invest Dermatol 121:862–868
Hearn RMR, Kerr AC, Rahim KF, Ferguson J, Dawe RS (2008) Incidence of skin cancers in 3867 patients treated with narrow-band ultraviolet B phototherapy Brit. J Dermatol 159:931–935
Huang CW, Cheng JY, Yen MH, Young TH (2009) Electrotaxis of lung cancer cells in a multiple-electric-field chip. Biosens Bioelectron 24:3510–3516
Imajoh M, Sugiura H, Oshima S (2004) Morphological changes contribute to apoptotic cell death and are affected by caspase-3 and caspase-6 inhibitors during red sea bream iridovirus permissive replication. Virology 322:220–230. doi:10.1016/j.virol.2004.02.006
Jhappan C, Noonan FP, Merlino G (2003) Ultraviolet radiation and cutaneous malignant melanoma. Oncogene 22:3099–3112
Kim JA, Ahn BN, Kong CS, Kim SK (2013) The chromene sargachromanol E inhibits ultraviolet A-induced ageing of skin in human dermal fibroblasts. Br J Dermatol 168:968–976
Kimura H et al (2010) UV light killing efficacy of fluorescent protein-expressing cancer cells in vitro and in vivo. J Cell Biochem 110:1439–1446
Krutmann J (2003) Premature skin aging by ultraviolet radiation and other environmental hazards. The molecular basis. Hautarzt 54:809
Kvam E, Dahle J (2004) Melanin synthesis may sensitize melanocytes to oxidative DNA damage by ultraviolet A radiation and protect melanocytes from direct DNA damage by ultraviolet B radiation. Pigm Cell Res 17:549–550
Lee E, Koo J, Berger T (2005) UVB phototherapy and skin cancer risk: a review of the literature. Int J Dermatol 44:355–360
Lo KY, Zhu Y, Tsai HF, Sun YS (2013) Effects of shear stresses and antioxidant concentrations on the production of reactive oxygen species in lung cancer cells. Biomicrofluidics 7, Article No. 064108
Lo KY, Wu SY, Sun YS (2016) A microfluidic device for studying the production of reactive oxygen species and the migration in lung cancer cells under single or coexisting chemical/electrical stimulation. Microfluid Nanofluid 20, Article No. 15
Miller ML, Andringa A, Elliott J, Conwell K 2nd, Dixon K, Carty MP (1998) The morphological and spectral phenotype of apoptosis in HeLa cells varies following exposure to UV-C and the addition of inhibitors of ICE and CPP32. Cell Prolif 31:17–33
Miyamura Y et al. (2008) The mechanism of melanin dispersion in human skin by ultraviolet (UV) radiation Pigm Cell Melanoma R 21:256–256
Moran DJ, Hollows FC (1984) Pterygium and ultraviolet radiation: a positive correlation. Br J Ophthalmol 68:343–346
Morris HA, O’Loughlin PD, Anderson PH (2010) Experimental evidence for the effects of calcium and vitamin D on bone: a review. Nutrients 2:1026–1035
Parisi AV, Green A, Kimlin MG (2001) Diffuse solar UV radiation and implications for preventing human eye damage. Photochem Photobiol 73:135–139
Picotto G, Liaudat AC, Bohl L, de Talamoni NT (2012) Molecular aspects of vitamin D anticancer activity. Cancer Invest 30:604–614
Reid IR, Bolland MJ, Grey A (2014) Effects of vitamin D supplements on bone mineral density: a systematic review and meta-analysis. Lancet 383:146–155
Saraiya M et al (2004) Interventions to prevent skin cancer by reducing exposure to ultraviolet radiation—a systematic review. Am J Prev Med 27:422–466
Situm M, Buljan M, Bulat V, Lugovic Mihic L, Bolanca Z, Simic D (2008) The role of UV radiation in the development of basal cell carcinoma. Coll antropol 32(Suppl 2):167–170
Stacey DW, Hitomi M, Kanovsky M, Gan L, Johnson EM (1999) Cell cycle arrest and morphological alterations following microinjection of NIH3T3 cells with Pur alpha. Oncogene 18:4254–4261. doi:10.1038/sj.onc.1202795
Sugita K, Koizumi K, Yoshida H (1992) Morphological reversion of sis-transformed NIH3T3 cells by trichostatin A. Cancer Res 52:168–172
Sun YS (2016) Comparison of chip inlet geometry in microfluidic devices for cell studies. Molecules 21 doi:10.3390/molecules21060778
Sun YS, Peng SW, Cheng JY (2012) In vitro electrical-stimulated wound-healing chip for studying electric field-assisted wound-healing process. Biomicrofluidics 6:34117. doi:10.1063/1.4750486
Tai MH, Weng CH, Mon DP, Hu CY, Wu MH (2012) Ultraviolet C irradiation induces different expression of cyclooxygenase 2 in NIH 3T3 Cells and A431 Cells: the roles of COX-2 are different in various cell lines. Int J Mol Sci 13:4351–4366
Wakamatsu K, Ito S (2006) Evaluation of melanin-related metabolites as markers of solar ultraviolet-B radiation. Pigm Cell Res 19:460–464
Wu SY, Hou HS, Sun YS, Cheng JY, Lo KY (2015) Correlation between cell migration and reactive oxygen species under electric field stimulation. Biomicrofluidics 9:054120. doi:10.1063/1.4932662
Wu SY, Sun YS, Cheng KC, Lo KY (2016) A wound-healing assay based on ultraviolet light ablation. J Lab Autom. doi:10.1177/2211068216646741
Yang F, Chen ZG, Pan JB, Li XC, Feng J, Yang H (2011) An integrated microfluidic array system for evaluating toxicity and teratogenicity of drugs on embryonic zebrafish developmental dynamics Biomicrofluidics 5, Article No. 024115
Yin L, Morita A, Tsuji T (2001) Skin aging induced by ultraviolet exposure and tobacco smoking: evidence from epidemiological and molecular studies. Photodermatol Photo 17:178–183
Acknowledgements
This work was financially supported by the Ministry of Science and Technology of Taiwan under Contract No. MOST 105-2112-M-030-002-MY2. The authors would like to thank Dr. Huai-Fang Chang for help in cell culturing.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Huang, CH., Hou, HS., Lo, KY. et al. Use microfluidic chips to study the effects of ultraviolet lights on human fibroblasts. Microfluid Nanofluid 21, 79 (2017). https://doi.org/10.1007/s10404-017-1922-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10404-017-1922-7