
Abstract
Electrokinetic transport of aqueous solutions containing multiple ionic species in surface charge governed nanofluidic flows has seen limited investigation with most experimental and modeling efforts emphasizing symmetric, monovalent electrolytes. In this work, numerical models coupling steady-state Poisson–Nernst–Planck and Stokes equations along with experimental investigations were developed to characterize electrokinetic transport of potassium phosphate buffer, containing K+, H2PO4 −, and HPO4 2− across positively charged nanocapillary array membranes with 10 nm diameter nanocapillaries, sandwiched between a source and permeate reservoir. While systematically increasing phosphate buffer concentration from 0.2 to 10 mM, 0.14 mM of methylene blue (MB) dye was added to the source reservoir to study the dominating transport mechanism under a potential bias (0–0.75 V). Experiments provided validation of numerical results that elucidate fundamental transport mechanisms as a function of ion type, buffer concentration, and externally applied potential. The nanocapillary exhibits permselectivity toward anions at lower buffer concentrations (0.2, 1 mM) and was more selective for HPO4 2− in comparison with H2PO4 −. Transport of K+, H2PO4 −, and HPO4 2− was dominated by electromigration, with negligible effects of diffusion and convection at all buffer concentrations. However, transport of MB+ was dominated by diffusion at 0.2 mM buffer concentration under all potential bias conditions. Significant effects of electromigration appeared at high potential biases (0.5–0.75 V) for 1 and 10 mM bulk buffer concentrations. Additionally, in the multicomponent ion system, at all concentrations, the vast majority of the current was carried by the phosphate buffer ions and not the MB ions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Transport of ions and molecules across nanoscale conduits forms an exciting and evolving area of scientific and technological investigation (Conlisk 2013; Prakash and Yeom 2014). In particular, nanochannels and nanocapillaries are two nanofluidic architectures used as functional transport conduits in a variety of emerging lab-on-chip devices for separation and detection (Howorka et al. 2001; Storm et al. 2005; Karnik et al. 2006; He et al. 2011), DNA and protein manipulation (Karnik et al. 2005, 2006; Plesa et al. 2013), molecular gating (Guan et al. 2011; Jin and Aluru 2011) and sieving (Luo et al. 1996), electrochemical reactors (Fu et al. 2007; Kim et al. 2009; Ma et al. 2013a, b), fundamental ion transport investigations (Jung et al. 2009; Guan et al. 2011; Swaminathan et al. 2012; Nandigana and Aluru 2012; Fleharty et al. 2014; Gamble et al. 2014), asymmetric nanoscale pumps (Siwy and Fulinski 2002), permselective membranes (Lee and Martin 2002; Yu et al. 2003), field effect switches (Fuest et al. 2015), water desalination (Kim et al. 2010; Prakash et al. 2014), and energy conversion devices (Pennathur et al. 2007; van der Heyden et al. 2007).
In most reports with polymeric or silica (SiO2) nanopores, including nanocapillary array membranes (NCAMs) (Kemery et al. 1998; Lee and Martin 2002; Prakash et al. 2007; Dekker 2007; Wang et al. 2009; Prakash et al. 2012), the applied streamwise or axial potential ranges within ±1 V with KCl being commonly used in the 10−2–3 M concentration range (Dekker 2007). However, studies using nanochannels, which are commonly fabricated in silicon or glass substrates, have explored a much broader concentration range for KCl, from ~10−7 to 1 M with applied streamwise or axial potentials on the order of several volts [~O(101)–O(102) V] (Karnik et al. 2005; Kim et al. 2007).
It is now generally accepted that conductance (the inverse of resistance to ionic flow and usually expressed as I/V, where I is the ionic current and V the potential) in nanofluidic architectures such as a nanopore, nanocapillary, or nanochannel is determined by surface charge at low electrolyte concentrations C bulk, with several previous reports identifying C bulk ~ 1 mM as the critical concentration for the transition from surface charge governed to bulk conductance behavior (Stein et al. 2004; Karnik et al. 2005; Fuest et al. 2015), where the conductance of the nanofluidic architecture is independent of electrolyte concentrations below C bulk ≤ 1 mM. In addition, several modeling studies have determined velocity and concentration profiles for ions and other species to explain observed trends in experimental data (Datta et al. 2009; Nandigana and Aluru 2012; Zambrano et al. 2012; Schnitzer and Yariv 2013), including dependence of surface charge on ion-adsorption-related fluctuations (Smeets et al. 2009).
By contrast, biological systems comprise electrolyte mixtures or multicomponent systems with a variety of ionic valences. Past research has discussed anomalous mole fraction effects, i.e., a nanochannel showing preferred binding selectivity for a particular ionic species in mixtures resulting in lower conductance compared to pure salts at same concentration leading to selectivity in negatively charged synthetic nanopores in the presence of multivalent cations (Gillespie et al. 2008). Moreover, it was recently also shown that the current rectification in nanocapillary membranes could be altered by modifying the degree of asymmetry between the cross-sectional areas of reservoir baths connecting the nanocapillary membranes for a potassium phosphate buffer (Wang et al. 2015).
Studies on DNA translocation through solid-state nanopores reveal that translocation time of DNA increases with decrease in cation size of surrounding bulk electrolyte (Kowalczyk et al. 2012). Previous modeling efforts have shown the effect on electroosmotic flow and velocity profiles with the addition of mono- and multivalent ions to aqueous NaCl in confined micro- and nanochannel systems (Conlisk et al. 2002). Capillary electrophoresis has also been used to estimate the empirically adjusted mobility of multivalent organic ions as a function of background electrolyte ionic strength (Friedl et al. 1995; Koval et al. 2005).
Despite these significant advances, several unanswered questions remain in completely understanding the physics of nanoscale transport, especially in heterogeneous systems that contain multiple ionic species with mixed valences. Specifically, unlike translocation events of DNA and other biomolecules across solid-state nanopores, which are typically quantified by blockage current measurements (Kowalczyk et al. 2012; Mathwig et al. 2015), translocation of smaller ionic analytes (e.g., dyes like methylene blue, fluorescein, or protein fragments) does not block net ionic current appreciably as the characteristic size of the molecule is still much smaller compared to the nanofluidic architecture (at least an order of magnitude or more physical dimension difference between molecule size and characteristic length scale of the nanofluidic architecture). Furthermore, the role of ion type on permselectivity in multicomponent systems is largely unknown. Quantitative understanding of such ionic transport in heterogeneous systems can provide pathways to fundamentally understand the functionality of biological systems such as ion channels which exhibit excellent selectivity in regulating transport of specific ions, despite being surrounded by extra-cellular fluid comprised of various types of ions and molecules (Siwy and Howorka 2010).
The main measurable parameter in nanofluidic electrokinetic transport experiments is the electric current, caused by the flux of ions J i (Prakash et al. 2008), as given by Eq. (1),
For a given species i, D i is the diffusion coefficient, c i is the concentration, \(\xi_{i}\) is the ionic mobility, z i is the ion valence, Φ is the potential, and u is the convective velocity. Ionic mobility is related to the diffusion coefficient as \(\xi_{i} = \frac{{eD_{i} }}{{k_{b} T}}\) where e is the elementary charge, k b is the Boltzmann constant, and T is the absolute temperature (Vlassiouk et al. 2008). The summation of the ionic flux (units: mol m−2 s−1) across all the species and subsequently integrating over the cross section area and multiplying with Faraday constant and valence of each species then provide the total ionic current, I which is typically measured. Equation (1) shows that there are three contributions to the total ionic flux and subsequently the ionic current, I. The first term represents the contribution from diffusion driven by a concentration gradient of species i, the second term is the contribution due to the electromigration of the species, and the third term is the flux arising due to the convective transport of ionic species in the bulk fluid. Given the complexity of most nanoscale transport models (Conlisk 2013), the convective flux is often considered minimal and neglected as the total flux is considered to be dominated by electromigration in low Reynolds number fluid flow (Jin et al. 2007; Vlassiouk et al. 2008; Guan et al. 2011). In some cases, it has been estimated that the convective flux contributes at most 20 % to the total flux, where the convective flux was found to increase with increase in wall surface charge and applied bias (Vlassiouk et al. 2008). Finally, the relative contribution of each of the three (diffusive, electromigration, and convective) components to total ionic flux has seen only limited modeling-based investigations.
In this paper, using a combination of both numerical modeling and experimental methods, electrokinetic transport of dilute aqueous potassium phosphate buffer with methylene blue dye at pH 7.0 ± 0.2 was studied across NCAMs with nominal diameter of 10 nm in the sub-1 V axial or streamwise applied potential range. Sub-1 V potentials were applied to minimize effects of concentration polarization (Kim et al. 2007) induced nonlinear electrokinetic flows and to mitigate any influence of faradaic reactions (Albrecht 2011). Therefore, the purpose of this paper is to elucidate the effects of electric double layers (EDLs) on nanocapillary selectivity due to ion type, nanocapillary conductance, and equilibrium concentration distributions while describing the role of each of the three transport contributory mechanisms in an electrolyte mixture comprising multivalent ions and a positively charged organic molecular dye.
2 Experimental setup
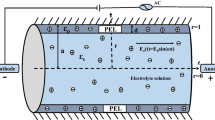
Ion transport measurements were performed on nuclear track-etched, PVP-coated polycarbonate NCAMs with a nominal nanocapillary diameter of 10 nm (GE Water & Process Technologies) and a total membrane diameter of 25 mm. NCAMs were pre-treated in DI water (Millipore, 18.2 MΩ) for 48 h and the working buffer solution for 4 h immediately prior to the experiments, following previously reported procedures (Vitarelli et al. 2011). Current measurement and application of voltages were performed using a Gamry Reference 600 potentiostat using a four-electrode setup in a standard chronoamperometry experiment (Bard and Faulkner 2004) with axial voltages stepped sequentially from 0 to 0.75 V between the source and permeate reservoirs (Fig. 1). The use of four electrodes minimizes measurement errors due to electrode polarization. Potassium phosphate buffer, comprising both the monobasic and dibasic phosphate anions (Sigma Aldrich, USA), at three buffer concentrations of 0.2, 1, and 10 mM and at pH 7.0 ± 0.2 was prepared as the working solution. The buffer provides control over electrolyte pH which is critical since surface charge is a major influence on nanoscale transport and pH changes can cause changes to the surface charge of the nanocapillary walls (Chun and Stroeve 2001; Wu et al. 2010).

a Schematic representation of the experimental setup. The source and the permeate reservoirs, connected by a nanocapillary array membrane, were filled with 500 ml potassium phosphate buffer with concentration varying from 0.2 to 10 mM at a pH of 7 ± 0.2. O-rings around the NCAMs ensure leak-free operation across the permeation cell. The potential electrodes (Ag/AgCl) and the current measurement electrodes (Au) were placed as shown. 0.14 mM of methylene blue dye was added to the source reservoir at the beginning of each experiment. b Schematic for the numerical model for the two-dimensional axisymmetric geometry used to numerically calculate ionic transport under an applied potential bias. The diameter of the nanocapillary and reservoirs was fixed at 10 and 200 nm, respectively. The surface charge of reservoir walls was −0.1 mC/m2, and the nanocapillary walls were assigned a positive surface charge of 0.35 mC/m2 based on previous reports (Jin et al. 2007)
Experiments were carried out in a custom cast acrylic permeation cell (Fig. 1a) with 500 mL solution of potassium phosphate buffer on each side. The permeation cell was placed in an earth grounded Faraday cage to minimize effects of external electrical noise on the electric current measurements. The potential difference across the NCAMs was applied through Ag/AgCl reference electrodes (Bioanalytical Systems) with current measured with Au electrodes away from the NCAMs as depicted schematically in Fig. 1a. The Ag/AgCl reference electrodes were connected to a high impedance input (10 GΩ) on the potentiostat, while both the working and counter electrodes were 1 mm diameter gold (Au) wires (99.9 % purity from Alfa Aesar) which had been cleaned using a standard RCA-1 cleaning process (100:10:1 solution of DI water, 30 % H2O2, and 28 % NH4OH at 73 °C for 10 min). Au electrodes were thoroughly rinsed with DI water, dried in filtered dry air, and stored in a sealed container till use (Prakash and Karacor 2011).
Methylene blue (MB) was added as a tracer dye and a representative small organic molecule to the solution on the source reservoir at a 0.14 mM concentration for all experiments (Fig. 1a). 0.14 mM of MB was added after developing calibration curves with sufficient signal to noise ratio for detection of MB in the permeate reservoir over a 1-h experimental period (Bellman 2011). For generating the calibration curves, absorptivity of each 3 ml sample was measured with a Thermo-Scientific Evolution 300 UV/Vis at 665 nm, which was translated to an equivalent concentration and subsequently current following a detailed, multipoint calibration of absorption intensity for known concentrations (Singhal and Rabinowitch 1967; Bellman 2011) (see supporting information for more discussion). In order to determine the steady-state flux of MB+, the measured concentration via UV/Vis was normalized by time-averaging the concentration at various extraction times.
Total current was recorded throughout the entire 3600 s experiment, while holding the potential difference across the electrodes and measuring the resulting current for each applied potential. Additionally, 3 ml sample of solution was extracted using a dedicated pipette from the permeation cell at 0, 8, 15, 30, 45, and 60 min after gently stirring the reservoirs to ensure a uniformly mixed bulk solution prior to sample extraction. It is important to note that the methylene blue ion is positively charged at the experimental conditions with unit valence (Chen et al. 2001).
3 Numerical model
3.1 Geometry
A numerical model for the experimental setup described above was also developed. The geometry of the nanocapillary and the connecting reservoirs were assumed to be cylinders as shown in Fig. 1b. In the experiments, the volume of reservoirs is significantly larger in comparison with the volume of the nanocapillary. However, identical match in numerical models is not possible given the tremendous requirement for computational resources for modeling a nanometer-scale transport system coupled to reservoirs with 500 mL volume. Therefore, for the numerical model, a scaling approach was used.
The diameter of the nanocapillary was 10 nm as in the experimental setup. The cylindrical reservoirs were set at a diameter of 200 nm and a height of 250 nm to allow a significantly larger reservoir volume with respect to the nanocapillary. In order to maintain an equivalent driving force for electrokinetic flow between the experiments and the numerical model, the length of the nanocapillary was chosen to be 500 nm (see further discussion in Sect. 3.2 below) for all the calculations reported here.
3.2 Governing equations and boundary conditions
The reservoirs were made from high purity cast acrylic, which is known to have surface charge properties of poly(methyl methacrylate) or PMMA with a low surface charge in comparison with polycarbonate (Kirby and Hasselbrink 2004; Conlisk 2013). Using known reports for PMMA (Conlisk 2013), the reservoirs were assigned a surface charge, σ w = −0.1 mC/m2 which is similar in value to other numerical studies (Jin et al. 2007) of systems with similar geometries and materials. The NCAMs used in this work are polycarbonate membranes coated with PVP (polyvinylpyrrolidone) (GE water & Process Technologies). It is known that for NCAMs, electrolyte pH and concentrations above 1 mM (Keesom et al. 1988) may cause changes in surface charge and/or zeta (ζ) potential due to ion adsorption or pH-dependent surface protonation and de-protonation of the surface terminal functional groups. In the absence of established reactions and known reaction coefficients, determination of accurate surface charge for the NCAMs remains an open question. However, based on past work at pH 7 (Kuo et al. 2001; Jin et al. 2007), an estimated surface charge, σ s = 0.35 mC/m2 was used for the models in the present work.
Under the influence of an applied potential bias across the nanocapillary, the electrokinetic flow of individual species (K+, MB+, H2PO4 −, and HPO4 2−), which comprise the phosphate buffer, is composed of the diffusive, electromigration, and convective fluxes as described by Eq. (1). The steady-state flux continuity is given by:
Dirichlet boundary conditions were imposed on the ionic concentration for K+, H2PO4 −, and HPO4 2−, at the inlet and outlet of the reservoir (C (inlet, outlet) = C bulk). MB+ concentration at the inlet of source reservoir was fixed to 0.14 mM, with no MB+ initially in the permeate side or outlet reservoir, matching the experimental conditions. No ionic flux normal to all the other nanocapillary and reservoir walls was permitted similar to previously reported models (Guan et al. 2011).
The equilibrium distribution of potential Φ, in the reservoirs and nanocapillary, is governed by the Poisson equation,
where ε 0 is the permittivity of free space, ε w (=80) is the relative permittivity of water and was assumed to be constant, and ρ s is the volumetric space charge density, as defined in Eq. (4). A Dirichlet boundary condition was also applied for the potential at the reservoirs, such that Φ at the inlet of source reservoir = V i , is same as the inlet axial potential of experimental conditions.
As briefly discussed above, modeling a scaled system was necessary for a reasonable computational load. Therefore, it is important to note that the length of the nanocapillary was 500 nm in the numerical model in comparison with the actual nanocapillary length of 6 µm in the experiments. The scaling in the models for a nanocapillary length of 500 nm for the same 10 nm diameter continues to preserve the near-infinite length of the nanocapillary compared to the diameter. However, this scaling poses an interesting challenge. Since the driving force in electrokinetic flows is the electric field in the axial or streamwise direction (Fig. 1), if the permeate reservoir in numerical model is grounded, it will not provide an equivalent driving force across the nanocapillary with respect to the experiments as the magnitude of electric field inside the simulated nanocapillary would then be 12 times higher than that of the experimental field (E z = −dΦ/dz, as field is inversely proportional to length of nanocapillary). Therefore, Φ at the source and permeate reservoir was set to V i and 11V i /12 (Fig. 1b), respectively, in the numerical model resulting in a potential difference of V i /12 across the nanocapillary. Subsequently, dividing the potential difference by the length of the nanocapillary (500 nm in model) results in a net electric field of V i /6 V/µm, thus matching the magnitude of electric field with experiments.
A Neumann boundary condition was applied to the reservoir walls (Eq. 5) and to the positively charged nanocapillary walls (Eq. 6) (Guan et al. 2011).
The bulk fluid transport (water) is governed by conservation of mass or velocity continuity (Eq. 7), and the fluid flow is described by Stokes equation (shown in Eq. 8) (Jin et al. 2007),
where µ is the dynamic viscosity of water at 293 K, p is the pressure, and −ρ s ∇Φ is the body force on bulk fluid due to space charge and electric field. The gage pressure was set to zero at both the inlet and outlet reservoirs, and a no-slip boundary condition was imposed on all other walls. A summary of parameters used in the numerical calculations is provided in Supporting Information (Table S1). Molecular dynamic simulations have shown that no-slip boundary condition is satisfied when considering electrokinetic transport of ions in 5.8 nm (Zhu et al. 2005) and 7 nm channels (Prakash et al. 2015). Therefore, the use of the continuum-based modeling approach and no-slip boundary condition for a 10 nm diameter nanocapillary is in line with existing methods reported for nanofluidics. Unlike ultra-fast flow of water observed in hydrophobic carbon nanotubes, polymer membranes, when coated with PVP, demonstrate an intrinsic membrane roughness and are hydrophilic. In such cases, no significant flow enhancement was observed, and the no-slip boundary condition can be assigned to characterize the overall transport (Majumder et al. 2005; Joseph and Aluru 2008).
The complete set of coupled equations described above, commonly known as Poisson–Nernst–Planck (PNP) and Stokes equations were solved simultaneously numerically with COMSOL Multiphysics version 4.4 for the entire reservoir–nanocapillary geometry. Solutions to these equations reported here were verified for mesh independence, and the relative tolerance for convergence was set to 10−6. The PNP–Stokes equations were solved in 2D cylindrical coordinates (r, z) with an axisymmetric boundary condition about the z-axis (r = 0), and therefore, the gradient of all quantities in the azimuthal coordinate Ψ was assumed to be zero (Fig. 1b). The assumption on the gradient in Ψ is valid for the nanocapillary here as all the boundary conditions are uniform in Ψ and symmetric about the z-axis. Solving the system of equations in three dimensions (using a 3D geometry of same radial and axial dimensions, not explicitly shown) confirmed the assumption that derivative of all quantities with respect to Ψ was negligible and does not affect the overall results reported here. It is essential to note that solving the same set of equations in 3D consumes significantly more computational resources. The net current through the nanocapillary of radius R was numerically calculated by integrating the total ionic flux through the nanocapillary cross section as shown in Eq. (9),
4 Results and discussion
4.1 Numerical model validation
The numerical model was validated by comparing the calculated current for all three electrolyte concentrations against the total measured experimental current across the 10 nm nanocapillary. Figure 2a shows the total current from experiments (open symbols) and numerical simulations (lines) as a function of the applied potential difference across the NCAM. The measured and numerically determined total current was in agreement within experimental uncertainty.

a Experimentally measured total current compared against the numerically calculated total current across the NCAM for an applied potential difference between 0 and 0.75 V, while the bulk potassium phosphate buffer concentration was increased from 0.2 to 10 mM. The current was found to increase linearly with inlet potential for a given buffer concentration. b Experimentally and numerically determined conductance as a function of buffer concentration shows reasonable agreement within experimental uncertainty. Though the conductance is found to increase linearly with buffer concentration for C bulk ≥ 1 mM, a similar scaling was not observed when the bulk buffer concentration was further reduced to 0.2 mM suggesting that the nanocapillary operates under surface charge governed regime as shown previously (Stein et al. 2004; Karnik et al. 2005; Fuest et al. 2015). Errors bars indicate one standard deviation from the mean
Numerical current was estimated by integrating the diffusive, electromigration, and convective flux of all four ionic species (K+, H2PO4 −, HPO4 2−, and MB+) across the cross-sectional area of the nanocapillary (Eq. 9). The numerical current reported in Fig. 2a accounted for the total number of nanocapillaries (~1.1 × 109 as estimated from manufacturer’s specification) to compare against the NCAM used in the experiment. Experimentally measured conductance and the calculated conductance were found to be in agreement within experimental uncertainty and were also found to follow the expected trends for nanoscale transport with decreasing electrolyte concentration (Karnik et al. 2005; Fuest et al. 2015; Fig. 2b).
In order to further validate the numerical model, Fig. S1 (Supporting Information) shows the concentration distribution across the reservoir–nanocapillary system at rest state (no potential gradient across the nanocapillary, though MB gradient exists between source and permeate reservoir). Specifically, Fig. S1 shows the axial (at r = 0 nm) and radial (at z = 500 nm) variation of concentration across the reservoir–nanocapillary system for all the three reservoir buffer concentrations. As expected (Jin et al. 2007), the centerline concentrations of K+, H2PO4 −, and HPO4 2− ions stay constant in the respective reservoirs. Inside the positively charged nanocapillary the concentration of co-ions (K+ and MB+) decrease and counter-ions (H2PO4 − and HPO4 2−) increase in comparison with their respective bulk concentration in order to maintain electroneutrality (Jin et al. 2007; Vlassiouk et al. 2008).
As the cross-sectional area of the reservoirs is 400 times larger than the nanocapillary in the numerical models, the reservoirs were equipotential with the entire applied potential drop occurring across the nanocapillaries also in agreement with previous results (Fuest et al. 2015). This linear potential drop across the nanocapillary (Fig. 3a) also shows that reservoir–nanocapillary system was not affected by polarization which is typically characterized by a rapid nonlinear potential decrease inside the nanocapillary (Vlassiouk et al. 2008). In addition, current scales linearly with potential as seen in Fig. 2a, an indication that the system is still in the ohmic region (Kim et al. 2007). From Fig. 3a, it is evident that the Donnan potential for a given concentration (e.g., plotted in Fig. S2a, supporting information) adds to the total potential at source reservoir/nanocapillary junction and is removed at the permeate reservoir/nanocapillary junction. Figure 3b shows the variation of average axial electric field (E z,avg) as a function of the applied inlet potential inside the nanocapillary, as calculated from the numerical models. The average axial electric field across the entire volume, β of the nanocapillary, is defined as \(\frac{1}{\beta }( {\iiint\limits_{\beta } {E_{z} }(r,z){\text{d}}\beta} )\), where E z (r, z) is the spatially dependent axial electric field inside the nanocapillary, also taking into account the entrance effects (Fig. S3) due to the Donnan potential. From Fig. 3b, the magnitude of E z,avg for a given inlet potential is similar in magnitude for all the test buffer concentrations and is seen to increase linearly with increase in inlet potential and is constant in magnitude inside the nanocapillary (Fig. S3).

a Variation of axial potential inside the reservoir and nanocapillary plotted for all the three buffer concentrations. Since the reservoir has a much higher cross-sectional area (~400 times) compared to that of nanocapillary in the numerical model, the resistance of the reservoir is negligible in comparison with that of nanocapillary (Fuest et al. 2015) and subsequently nearly the entire potential drop occurs across the length of the nanocapillary as depicted. The increase in potential at source reservoir/nanocapillary junction is due to the addition of Donnan potential to the bulk potential. b Numerically calculated average axial electric field E z, avg inside the nanocapillary as a function of applied inlet potential. The electric field varied linearly with increase in inlet potential
4.2 Species dependence on ionic current
The total measured current is a net summation of current contributions from all ionic species in solution. However, the experimental measurements from chronoamperometry cannot quantitatively distinguish between the current from various ionic species present within the electrolyte solution. Figure 4 shows the numerically calculated current across a single nanocapillary as predicted by the numerical model when V i was varied from 0 to 0.75 V for the three buffer concentrations of 0.2 mM (Fig. 4a), 1 mM (Fig. 4b), and 10 mM (Fig. 4c), respectively.

Numerically estimated current contributions from individual ions (K+, H2PO4 −, HPO4 2−, and MB+) to the total current when the buffer concentration was a 0.2 mM, b 1 mM, and c 10 mM, for a single nanocapillary. Results indicate that anionic current contributes to the majority of measured current at lower buffer concentrations (0.2, 1 mM), as expected due to the exclusion of co-ions from the positively charged nanocapillary under conditions of interacting double layers. Interestingly, the divalent anion was found to carry the larger fraction of current compared to the monovalent anion. d Selectivity of the nanocapillary, defined as the ratio of difference between the anionic current from cationic current to the total current as a function of buffer concentration. Due to near-perfect shielding of co-ions at 0.2 mM with a Debye length of 16.3 nm, selectivity was maximum with majority of cations excluded from the nanocapillaries. As the buffer concentration increases to 10 mM, NCAM selectivity decreased to allowing significantly more cation transport
At a buffer concentration of 0.2 mM (Fig. 4a), the current contribution of the divalent anion, HPO4 2−, was calculated to be 76 % of the total current. At 0.2 mM, the Debye length (Conlisk 2013) for the electrolyte solution was 16.3 nm, corresponding to overlapped EDLs for a 10 nm diameter nanocapillary. Under these conditions, the contribution of K+ to the total current was only ~6 %, implying high permselectivity of counter-ions in the positively charged nanocapillary. At C bulk = 1 mM, the contribution of the K+ current to total current increased to 32 % of the total current and was nearly two times the current arising from the monovalent anion, H2PO4 −. However, the total current was still dominated by the divalent anion (HPO4 2−), now contributing nearly half of the total current. As buffer concentration was further increased to 10 mM with a corresponding Debye length <3 nm, the magnitude of the current due to K+ was significantly higher (58 % of total current) compared to both HPO4 2− (28 % of total current) and H2PO4 − (13 % of total current) as shown in Fig. 4c.
The contribution of the other cation in solution, MB+, to the total current was the smallest in all cases compared to K+, H2PO4 − and HPO4 2−, with ~0.4 % of total current at C bulk = 10 mM, increasing to 3 % at C bulk = 1 mM, and to 4 % of the total current at C bulk = 0.2 mM. A comparison of the relative contributions to the current for each type of ion for the transport mechanisms described with Eq. (1) confirmed past known general trends hold even in the sub-1 V range for axial potential across long nanocapillaries; electromigration was the dominating transport mechanism for K+, H2PO4 −, and HPO4 2−, contributing to more than 99 % of the total current over combined contributions from convection and diffusion (Gillespie et al. 2008; Vlassiouk et al. 2008; Jin and Aluru 2011). It is noteworthy that MB+ did not follow these same trends with details on transport of methylene blue discussed subsequently.
As is well known, that decreasing electrolyte concentration increases Debye length and the EDL begins to overlap or interact within nanocapillary causing selective ion transport (Nishizawa et al. 1995). The nanocapillaries show permselectivity due to dominance of surface charge (Martin et al. 2001) and hence permit preferential counter-ion transport while excluding the co-ions (K+, MB+) to the surface charge polarity. Therefore, at the test concentrations with C bulk at 0.2 and 1 mM, it would be expected that the nanocapillaries would demonstrate preference for anions while excluding cations. In order to quantify the selectivity of the NCAMs here, a selectivity parameter S (Vlassiouk et al. 2008) was defined for the positively charged nanocapillary as the ratio of the difference in current carried by the majority anionic carriers and minority cationic carriers (K+, MB+) to the total measured ionic current,
The value S = 1 implies that the nanocapillary is perfectly permselective and the total current is carried only by counter-ions. Similarly, S = 0 implies that the current carried by majority anionic carriers and minority cationic carriers are equal in magnitude. For the range of applied potentials (0.1–0.75 V), the selectivity was estimated to be 0.79 ± 0.05, 0.30 ± 0.03, and −0.18 ± 0.01 for C bulk = 0.2, 1, and 10 mM, respectively, as shown in Fig. 4d. Given that K+ has the highest ionic mobility among electrolyte ions (Table S1) and also is poorly screened at C bulk = 10 mM (Fig. S1a), the dominating electromigration flux of K+ (Eq. 1) at C bulk = 10 mM contributes to 58 % of the total current resulting in negative selectivity (−0.18 ± 0.01). It is important to note that the selectivity remains constant (Fig. 4d) for a given buffer concentration despite the increase in electric field. In order to observe a decline in S, which indicates polarization effects, an electric field of ~1 MV/m would be needed, which is an order of magnitude higher than the electric field applied in this work (Fig. 3b) (Vlassiouk et al. 2008). Previous results have shown that when nanocapillary length is >100 nm, then change in selectivity due to interacting Donnan fields is mitigated, especially for the concentration ranges considered in this study (Vlassiouk et al. 2008). Finally, even at 0.2 mM, with highly overlapped double layers for the nanocapillary, perfect permselectivity was not attained, indicating that a small fraction of current is still carried by the cations.
4.3 Methylene blue transport characterization
Total current measurements from chronoamperometry experiments and from the numerical models provided information on the extent of selective ion transport of the NCAMs under varying bulk phosphate buffer concentration. In order to experimentally quantify the selective ion transport inside the NCAM in heterogeneous systems, positively charged MB+ dye at a concentration of 0.14 mM was added to the source side (Fig. 1), and the time-averaged MB+ mass transport was measured via UV/Vis as discussed in the experimental methods section. The axial flux of MB+ and J +MB provides a quantification of the current carried by MB+.
The transport of MB+ inside the nanocapillary is also governed by the coupled PNP–Stokes’ equation as discussed in the introduction and the methods sections above. Figure 5a shows a qualitative schematic of the direction of individual driving forces for MB+ leading to a net flux. As the positively charged nanocapillary has a net resultant negative space charge density (Supporting Information, Fig. S4a and S4b), the net electroosmotic flow (EOF) under the application of a positive bias is toward the positive electrode as shown in the schematic. Therefore, the convective component of current for MB+ is always negative and becomes more negative with increase in applied bias. The diffusive component of current always drives MB+ in the direction of concentration gradient, which is from the source reservoir to permeate reservoir, resulting in a positive flux. The electromigration component of the current drives MB+ (and will also have the same effect on K+) toward the permeate reservoir and is proportional to the product of average concentration of MB+ and the axial electric field inside the nanocapillary resulting in positive flux.

a Qualitative schematic depicting net transport of the MB+ ion inside the NCAM showing the direction of convective, diffusive, and electromigrative fluxes during transport across the nanocapillaries. Methylene blue (MB+) current is determined experimentally from the flux of the MB dye measured via UV/Vis spectroscopy and compared against numerically estimated MB+ current estimated for buffer concentration of b 0.2 mM, c 1 mM, d 10 mM, respectively. Lack of quantitative agreement at specific instances (e.g., 0.75 V and 0.2 mM), despite similarity in qualitative trends for MB+ current, in numerical and experimental currents is discussed in the main text. Also, for a given potential, numerically calculated methylene blue current was found to decrease with reduction in buffer concentration from 10 to 0.2 mM, as also observed in the experimental data
The experimentally measured time-averaged MB+ mass transport (units: \({\text{mol}}\,{\text{s}}^{ - 1}\)) was converted to an equivalent current by multiplying with Faraday constant and its valence (which is unity) and is shown in Fig. 5b–d. Experimental current and the variation in data represented by standard deviation was obtained with at least three runs for the same conditions over multiple membranes, relying on previously reported procedures to allow for experimental repeatability (Bellman 2011; Vitarelli et al. 2011). Uncertainties in the experimental NCAM system include a 10 % uncertainty in the length (Jin et al. 2007), tolerance of 20 and 10 % in the radius and NCAM number density, respectively (Vitarelli et al. 2011). These geometric uncertainties were taken into account and quantified in the numerical models as the electric field acting within the nanocapillary is inversely proportional to the length of the nanocapillary, and hence, a shorter capillary will lead to a higher driving electric field and a higher measured ionic flux. Similarly, an enlarged pore would also permit a higher flux and once again the spectroscopically determined current would be higher than the numerically estimated current for MB+.
In the 0–0.25 V applied potential range, the numerically calculated and experimentally determined current agree within experimental error for all conditions. However, as the applied potential was increased to 0.5 V, the numerically predicted current agreed well with measured current for C bulk = 10 and 1 mM but was lower than the UV/Vis measured current for C bulk = 0.2 mM. Furthermore, as the applied potential was increased to 0.75 V, the numerically determined current for MB+ is under predicted compared to the experimental MB+ current for C bulk = 1 and 0.2 mM. Now, the question arises as to why the numerical model does not match experimental data for these specific instances, even though the qualitative trends are similar and quantitative agreement exists at all other tested conditions?
One possible reason for this additional experimental flux at lower buffer concentrations (1 and 0.2 mM) and higher applied potentials (0.75 and 0.5 V) could arise due to complex flow reversal at reservoirs (Fig. S5), which are also charged. Switch in polarity of surface charge from positive (at the nanocapillary walls) to negative (in the reservoir) can also induce local flow reversal near the reservoir–nanocapillary junction due to local space charge inversion though the overall flow satisfies conservation of mass in the complete nanocapillary–reservoir system. Since NCAMs in the present study contain a very high density of nanocapillaries (1.1 × 109 ± 0.1 × 109 nanocapillaries), flow reversal at reservoir–nanocapillary junction could be further enhanced than predicted by the numerical model (where only one nanocapillary is considered) thus increasing the overall MB+ flux. It is possible that such flow reversal at the nanocapillary–reservoir interface can potentially cause localized concentration gradients, which are not captured in the steady-state numerical model. Recent studies demonstrate that such concentration gradients can actually cause surface charge inversion inside nanofluidic systems governed by counter-ion adsorption, especially with multivalent ions which could also potentially enhance MB+ transport (Li et al. 2015) but are beyond the computational scope of this work.
It is also worth noting that the experimentally measured MB+ current did not increase linearly with applied potential as was the trend with the other ions, K+, H2PO4 −, and HPO 24 . One could hypothesize that the current trends for the MB+ ions could possibly be explained by blockage current effects, similar to DNA translocation studies in a 10 nm nanocapillary (Mathwig et al. 2015); however, no evidence of blockage currents was observed in the experimental data, thus reinforcing the applicability of continuum-based modeling for the present study. Therefore, the observed trends in transport were further analyzed based on the three mechanisms of ion transport as summarized in Eq. (1) above.
Figure 6 shows the numerically calculated components of diffusion, electromigration, and convection currents to the total contribution for MB+ for a single nanocapillary as a function of inlet potential for the three buffer concentrations. The contribution of convection toward total MB+ current is negligible for all three tested buffer concentrations, with velocity profile plotted in Fig. S6 (supporting information). One numerical consequence is that the coupled PNP–Stokes equation can now be solved without including the Stokes equation, which could potentially decrease computational time and resources without significantly affecting the results. Furthermore, it was observed that for all three buffer concentrations (0.2, 1, and 10 mM; Fig. 6a, c), diffusion of MB+ cannot be neglected with respect to the electromigration of MB+. For instance, at the lowest buffer concentration of 0.2 mM, diffusion current was always higher than the electromigration current, regardless of the applied external potential.

Contributions of diffusion, electromigration, and convection for MB+ transport across a single nanocapillary for a buffer concentration of a 10 mM, b 1 mM, and c 0.2 mM, as calculated from numerical models. Data are plotted for a nanocapillary surface charge density of 0.35 mC/m2 (Jin et al. 2007). MB+ transport unlike that of K+, H2PO4 −, HPO4 2−, and HPO4 2− is dominated by diffusion especially at lower buffer concentrations with effects of electromigration contributing only at higher inlet potential and higher buffer concentrations. Values plotted for the geometric profile represented in Fig. 1b
At the highest phosphate buffer concentration of 10 mM, the diffusion component of the current was greater than the electromigration term for an applied potential of 0.25 V and both diffusion and electromigration have equivalent contributions at 0.5 V, with electromigration beginning to dominate at 0.75 V. Therefore, depending on the applied potential bias and C bulk, the actual dominating transport mechanism of MB+ across the nanocapillary was altered.
5 Summary and conclusions
Numerical calculations using coupled Poisson–Nernst–Planck and Stokes equations along with experiments were conducted to quantify ion transport for monovalent and divalent ions in a multicomponent electrolyte solution (K+, H2PO4 −, and HPO4 2−and methylene blue) across 10 nm nominal diameter positively charged nanocapillary array membranes. PNP–Stokes’ equations were shown to be accurate in quantifying transport mechanisms of both the bulk buffer and trace dye analytes in the present work. Numerical models matched experimentally measured current within experimental error and provided a quantitative description of the nanocapillary as function of Debye length and subsequently the electric double layers. It was found that the transport of buffer ions, K+, H2PO4 −, and HPO4 2− was dominated by electromigration while the added dye, MB+ shows a strong dependence on bulk buffer concentration with the diffusive flux dominating at lower applied potentials and at lower buffer concentration (C bulk ≤ 1 mM). Also, the nanocapillary membranes exhibit permselectivity toward anions and with the divalent ions carrying the bulk of the ionic current at buffer concentration ≤1 mM. The MB+ contribution was <5 % of the total current for all conditions evaluated here, in contrast to the current carried by the ions comprising the phosphate buffer solution. Given the nanocapillary length scales along with the use of several ions relevant to biological media, this work may provide insight to fundamental transport mechanisms in a variety of biological systems and complex miniature analytical systems.
References
Albrecht T (2011) How to understand and interpret current flow in nanopore/electrode devices. ACS Nano 5(8):6714–6725
Bard A, Faulkner L (2004) Electrochemical methods: fundamentals and applications. Wiley, Hoboken
Bellman KL (2011) Identification of low potential onset of concentration polarization and concentration polarization mitigation in water desalination membranes. The Ohio State University, Columbus
Chen J, Cesario TC, Rentzepis PM (2001) Effect of pH on methylene blue transient states and kinetics and bacteria photoinactivation. J Phys Chem A 115:2702–2707
Chun K-Y, Stroeve P (2001) External control of ion transport in nanoporous membranes with surface modified self-sssembled monolayers. Langmuir 17:5271–5275
Conlisk AT (2013) Essential of micro-and nanofluidics. Cambridge University Press, New York
Conlisk AT, McFerran J, Zheng Z, Hansford D (2002) Mass Transfer and flow in electrically charged micro- and nanochannels. Anal Chem 74(9):2139–2150
Datta S, Conlisk AT, Li HF, Yoda M (2009) Effect of divalent ions on electroosmotic flow in microchannels. Mech Res Commun 36(1):65–74
Dekker C (2007) Solid-state nanopores. Nat Nanotechnol 2:209–215
Fleharty ME, van Swol F, Petsev DN (2014) The effect of surface charge regulation on conductivity in fluidic nanochannels. J Colloid Interface Sci 416(15):105–111
Friedl W, Reijenga JC, Kenndler E (1995) Ionic strength and charge number correction for mobilities of multivalent organic anions in capillary electrophoresis. J Chromatogr A 709(1):163–170
Fu J, Schoch RB, Stevens AL, Tannenbaum SR, Han J (2007) A patterned anisotropic nanofluidic sieving structure for continuous-flow separation of DNA and proteins. Nat Nanotechnol 2(2):121–128
Fuest M, Boone C, Rangharajan KK, Conlisk AT, Prakash S (2015) A three-state nanofluidic field effect switch. Nano Lett 15(4):2365–2371
Gamble T, Decker K, Plett TS, Pevarnik M, Pietschmann J-F, Vlassiouk I, Aksimentiev A, Siwy ZS (2014) Rectification of ion current in nanopores depends on the type of monovalent cations: experiments and modeling. J Phys Chem C 118(18):9809–9819
Gillespie D, Boda D, He Y, Apel P, Siwy ZS (2008) Synthetic nanopores as a test case for ion channel theories: the anomalous mole fraction effect without single filing. Biophys J 95(2):609–619
Guan W, Fan R, Reed MA (2011) Field-effect reconfigurable nanofluidic ionic diodes. Nat Commun 2(1):1–8
He M, Novak J, Julian BA, Herr AE (2011) Membrane-assisted online renaturation for automated microfluidic lectin blotting. J Am Chem Soc 133(49):19610–19613
Howorka S, Cheley S, Bayley H (2001) Sequence-specific detection of individual DNA strands using engineered nanopores. Nat Biotechnol 19:636–639
Jin X, Aluru NR (2011) Gated transport in nanofluidic devices. Microfluid Nanofluid 11(3):297–306
Jin X, Joseph S, Gatimu EN, Bohn PW, Aluru NR (2007) Induced electrokinetic transport in micro-nanofluidic interconnect devices. Langmuir 23:13209–13222
Joseph S, Aluru NR (2008) Why are carbon nanotubes fast transporters of water? Nano Lett 8(2):452–458
Jung J-Y, Joshi P, Petrossian L, Thornton TJ, Posner JD (2009) Electromigration current rectification in a cylindrical nanopore due to asymmetric concentration polarization. Anal Chem 81:3128–3133
Karnik R, Fan R, Yue M, Li D, Yang P, Majumdar A (2005) Electrostatic control of ions and molecules in nanofluidic transistors. Nano Lett 5(5):943–948
Karnik R, Castelino K, Majumdar A (2006) Field-effect control of protein transport in a nanofluidic transistor circuit. Appl Phys Lett 88:123114
Keesom WH, Zelenka RL, Radke CJ (1988) A Zeta-potential model for ionic surfactant adsorption on an ionogenic hydrophobic surface. J Colloid Interface Sci 125(2):575–585
Kemery PJ, Steehler JK, Bohn PW (1998) Electric field mediated transport in nanometer diameter channels. Langmuir 14(10):2884–2889
Kim SJ, Wang Y-C, Lee JH, Jang H, Han J (2007) Concentration polarization and nonlinear electrokinetic flow near a nanofluidic channel. Phys Rev Lett 99(4):044501
Kim B, Yang J, Gong M, Flachsbart BR, Shannon MA, Bohn PW, Sweedler JV (2009) Multidimensional separation of chiral amino acid mixtures an a multilayered three dimensional hybrid microfluidic/nanofluidic device. Anal Chem 81:2715–2722
Kim SJ, Ko SH, Kang KH, Han J (2010) Direct seawater desalination by ion concentration polarization. Nat Nanotechnol 5(4):297–301
Kirby BJ, Hasselbrink EF (2004) Zeta potential of microfluidic substrates: 2. Data for polymers. Electrophoresis 25(2):203–213
Koval D, Kašička V, Zusková I (2005) Investigation of the effect of ionic strength of Tris-acetate background electrolyte on electrophoretic mobilities of mono-, di-, and trivalent organic anions by capillary electrophoresis. Electrophoresis 26(17):3221–3231
Kowalczyk SW, Wells DB, Aksimentiev A, Dekker C (2012) Slowing down DNA translocation through a nanopore in lithium chloride. Nano Lett 12(2):1038–1044
Kuo TZ, Sloan LA, Sweedler JV, Bohn PW (2001) Manipulating molecular transport through nanoporous membranes by control of electrokinetic flow: effect of surface charge density and debye length. Langmuir 17(20):6298–6303
Lee SB, Martin CR (2002) Electromodulated molecular transport in gold-nanotube membranes. J Am Chem Soc 124:11850–11851
Li SX, Guan W, Weiner B, Reed MA (2015) Direct observation of charge inversion in divalent nanofluidic devices. Nano Lett 15(8):5046–5051
Luo F, Giese CF, Gentry WR (1996) Direct measurement of the size of the helium dimer. J Chem Phys 104(3):1151–1154
Ma C, Contento NM, Gibson LR II, Bohn PW (2013a) Recessed ring-disk nanoelectrode arrays integrated in nanofluidic structures for selective electrochemical detection. Anal Chem 85:9882–9888
Ma C, Contento NM, Gibson LR II, Bohn PW (2013b) Redox cycling in nanoscale recessed ring-disk electrode arrays for enhanced electrochemical sensitivity. ACS Nano 7:5483–5490
Majumder M, Chopra N, Andrews R, Hinds BJ (2005) Nanoscale hydrodynamics: enhanced flow in carbon nanotubes. Nature 438(7064):44
Martin CR, Nishizawa M, Jirage K, Kang M, Lee SB (2001) Controlling ion-transport selectivity in gold nanotubule membranes. Adv Mater 13(18):1351–1362
Mathwig K, Albrecht T, Goluch ED, Rassaei L (2015) Challenges of biomolecular detection at the nanoscale: nanopores and microelectrodes. Anal Chem 87(11):5470–5475
Nandigana VVR, Aluru NR (2012) Understanding anomalous current–voltage characteristics in microchannel–nanochannel interconnect devices. J Colloid Interface Sci 384(1):162–171
Nishizawa M, Menon VP, Martin CR (1995) Metal nanotubule membranes with electrochemically switchable ion-transport selectivity. Science 268(5211):700–702
Pennathur S, Eijkel JCT, van den Berg A (2007) Energy conversion in microsystems: is there a role for micro/nanofluidics? Lab Chip 7:1234–1237
Plesa C, Kowalczyk SW, Zinsmeester R, Grosberg AY, Rabin Y, Dekker C (2013) Fast translocation of proteins through solid state nanopores. Nano Lett 13(2):658–663
Prakash S, Karacor MB (2011) Characterizing stability of “click” modified glass surfaces to common microfabrication conditions and aqueous electrolyte solutions. Nanoscale 3(8):3309–3315
Prakash S, Yeom J (2014) Nanofluidics and microfluidics: systems and applications. Elsevier, New York
Prakash S, Yeom J, Jin N, Adesida I, Shannon MA (2007) Characterization of ionic transport at the nanoscale. Proce IMechE J Nanoeng Nanosyst 220:45–52
Prakash S, Piruska A, Gatimu EN, Bohn PW, Sweedler JV, Shannon MA (2008) Nanofluidics: systems and applications. IEEE Sens J 8(5):441–450
Prakash S, Pinti M, Bellman K (2012) Variable cross-section nanopores fabricated in silicon nitride membranes using a transmission electron microscope. J Micromech Microeng 22(6):067002
Prakash S, Shannon MA, Bellman K (2014) Water desalination: emerging and existing technologies. In: Reisner DE, Pradeep T (eds) AquaNanotechnology. CRC Press, pp 533–562
Prakash S, Zambrano H, Fuest M, Boone C, Rosenthal-Kim E, Vasquez N, Conlisk AT (2015) Electrokinetic transport in silica nanochannels with asymmetric surface charge. Microfluid Nanofluid 19(6):1455–1464
Schnitzer O, Yariv E (2013) Electric conductance of highly selective nanochannels. Phys Rev E 87:054301
Singhal GS, Rabinowitch E (1967) Changes in the absorption spectrum of methylene blue with pH. J Phys Chem 71(10):3347–3349
Siwy Z, Fulinski A (2002) Fabrication of a synthetic nanopore ion pump. Phys Rev Lett 89(19):198103
Siwy ZS, Howorka S (2010) Engineered voltage-responsive nanopores. Chem Soc Rev 39(3):1115–1132
Smeets RMM, Dekker NH, Dekker C (2009) Low frequency noise in solid state nanopores. Nanotechnology 20:095501
Stein D, Kruithof M, Dekker C (2004) Surface charge governed ion transport in nanofluidic channels. Phys Rev Lett 93(3):035901-1–035901-4
Storm AJ, Storm C, Chen J, Zandbergen H, Joanny J-F, Dekker C (2005) Fast DNA translocation through a solid-state nanopore. Nano Lett 5(7):1193–1197
Swaminathan VV, Gibson II LR, Pinti M, Prakash S, Bohn PW, Shannon MA (2012) Ionic transport in nanocapillary membrane systems. J Nanopart Res 14(8):1–15
van der Heyden FHJ, Bonthuis DJ, Stein D, Meyer C, Dekker C (2007) Power generation by pressure-driven transport of ions in nanofluidic channels. Nano Lett 7(4):1022–1025
Vitarelli MJ, Prakash S, Talaga DS (2011) Determining nanocapillary geometry from electrochemical impedance spectroscopy by using a variable topology network circuit model. Anal Chem 83(2):533–541
Vlassiouk I, Smirnov S, Siwy Z (2008) Ionic selectivity of single nanochannels. Nano Lett 8(7):1978–1985
Wang Z, King TL, Branagan SP, Bohn PW (2009) Enzymatic activity of surface-immobilized horseradish peroxidase confined to micrometer-nanometer-scale structures in nanocapillary array membranes. Analyst 134:851–859
Wang H, Nandigana VVR, Jo KD, Aluru NR, Timperman AT (2015) Controlling the ionic current rectification factor of a nanofluidic/microfluidic interface with symmetric nanocapillary interconnects. Anal Chem 87(7):3598–3605
Wu Y, Misra S, Karacor MB, Prakash S, Shannon MA (2010) Dynamic response of AFM cantilevers to dissimilar functionalized silica surfaces in aqueous electrolyte solutions. Langmuir 26(22):16963–16972
Yu S, Lee SB, Martin CR (2003) Electrophoretic protein transport in gold nanotube membranes. Anal Chem 75:1239–1244
Zambrano H, Pinti M, Conlisk AT, Prakash S (2012) Electrokinetic transport in a water–chloride nanofilm in contact with a silica surface with discontinuous charged patches. Microfluid Nanofluid 13(5):735–747
Zhu W, Singer SJ, Zheng Z, Conlisk AT (2005) Electro-osmotic flow of a model electrolyte. Phys Rev E 71:041501
Acknowledgments
The authors would like to acknowledge partial financial support from the Defense Advanced Research Projects Agency (DARPA) administered through the Army Research Office (ARO) Grant Nos. W911NF09C0079, NSF-NSEC for the Affordable Nanoengineering of Polymeric Biomedical Devices EEC-0914790. In addition, partial financial support from NSF through grant number CBET-1335946 and the computational resources from the Ohio Supercomputer Center is also acknowledged. M. Fuest would like to thank NSF GRFP for financial support. We thank Ms. Karen Bellman, Ms. Wenqin He, and Mr. Cameron Bodenschatz for assistance with data collection and Dr. Emily Rosenthal-Kim for assistance in manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rangharajan, K.K., Fuest, M., Conlisk, A.T. et al. Transport of multicomponent, multivalent electrolyte solutions across nanocapillaries. Microfluid Nanofluid 20, 54 (2016). https://doi.org/10.1007/s10404-016-1723-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10404-016-1723-4