
Abstract
Extremely rapid water flow through carbon nanotubes has been observed in both experiment and simulation which has led to the suggestion that this material be used in a number of filtration applications. However, there is significant disparity in the magnitude of water permeability and the degree of flow enhancement compared with conventional porous materials in the literature. Here, we show that one of the causes of the disparity in simulation data is the variety of methods used to control temperature in molecular simulations. Not only can the choice of thermostat alter the flow rate and permeability by as much as five times, but it can determine whether the transport is observed to be frictionless or not. In addition to helping explain the disparate simulation results on transport in nanomaterials, this work provides some guidelines to help designing and interpreting molecular simulations of mass transport.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Carbon nanotubes have been suggested for use in a vast number of applications including the desalination of sea water; the removal of dangerous contaminants from water supplies; the separations of gases, ions and biomolecules; the sequencing of DNA in electrical devices and biosensors; and in nanofluidic devices. All of the listed uses involve flowing liquids or gases through the interior of the nanotubes to make use of the highly regular structure of the pores and their unusual transport properties. Water has been shown to flow through nanotubes with very high fluxes, in both experimental (Majumder et al. 2005; Holt et al. 2006) and simulation studies, (Hummer et al. 2001; Kalra et al. 2003; Corry 2008; Joseph and Aluru 2008; Thomas and McGaughey 2008; Song and Corry 2009; Thomas et al. 2010; Corry 2011; Ritos et al. 2014) with the interior of the nanotube suggested to provide a near-frictionless surface to water permeation. However, the predicted flow rates in the tubes differ by more than an order of magnitude amongst the many simulation studies, which is significant given that the magnitude of the water flux is essential to many of the suggested applications. Furthermore, while some studies claim no influence of nanotube length on the flow rate of water and infer frictionless flow (Kalra et al. 2003; Corry 2008; Nicholls et al. 2012a), other studies suggest decreasing flow rates with pore length (Su and Guo 2012). If molecular dynamics simulations are to be used to guide and understand the development of nanotube based technologies, then it is important that these disparate simulation results be reconciled.
One factor that may partly explain the different results from these studies is the distinct ways in which temperatures are controlled in these simulations. Usually simulations are compared to experimental conditions in which the average temperature of the system is constant. However, unmodified simulations yield a microcanonical ensemble in which the temperature is not controlled, but rather the number of atoms N, volume V and energy of the system E is maintained. Thus, simulations will typically modify the dynamics of the system with a thermostating algorithm to control the simulation temperature; but different algorithms can yield differing dynamics (Hunenberger 2005; Stoyanov and Groot 2005; Rosta et al. 2009). While some of the published simulations examining flow rates in carbon and other nanotubes employ a Berendsen thermostat (Hummer et al. 2001; Kalra et al. 2003; Peter and Hummer 2005; Won et al. 2006; Thomas and McGaughey 2008; Thomas et al. 2010; Nicholls et al. 2012a, b), other use a Nose–Hoover (Won and Aluru 2007; Joseph and Aluru 2008; Won and Aluru 2008; Khademi and Sahimi 2011; Cohen-Tanugi and Grossman 2012; Su and Guo 2012; Kannam et al. 2013), Langevin (Corry 2008; Hilder et al. 2009; Song and Corry 2009; Hilder et al. 2010; Corry 2011; Majumder and Corry 2011) or Gaussian (Liu et al. 2006) thermostat. A number of other studies do not specify how the temperature is controlled (Zhu and Schulten 2003; Li et al. 2007; Gong et al. 2008; Suk et al. 2008; Gong et al. 2010).
The precise influence of thermostating methods on the outcomes of molecular simulations is still not well understood, but previous studies have shown that temperature control algorithms can alter the dynamics of the system with respect to the microcanonical ensemble by, for example, damping the diffusion and rotational motion of molecules in the simulation. Furthermore, it has also been shown that different algorithms that adjust particle velocities in different ways can give significantly different dynamic properties (Basconi and Shirts 2013; Krishnan et al. 2013). In particular, it has been shown that the self-diffusivity, rotational correlation time and shear viscosity of water can all be altered by the thermostat choice (Basconi and Shirts 2013). While these studies examine the dynamics of liquids and solutes under equilibrium conditions, to our knowledge no systematic study has been made to examine the influence of temperature control algorithm in simulations of mass transport, in which there is directional flow of particles. Given the enormous interest in understanding mass transport in biological and synthetic nanoscale pores, we believe it is essential to see how different thermostating algorithms alter the simulated transport properties.
Here, we show that the water permeability and fluxes through carbon nanotube membranes found in molecular dynamics simulations are highly dependent on the thermostat that is used and that thermostat choice can have profound implications on whether friction is found within the flow. Not only does this help to explain some of the discrepancies amongst the published simulation data, it also provides a benchmark to aid in choosing the most appropriate thermostat in future simulations of mass transport.
2 Theoretical background
A large number of methods have been developed to control the temperature in molecular dynamics simulations. Rather than give a complete introduction to these here, we provide a summary of some of the differences critical to understanding this work and refer the reader to some excellent previous publications for further details. (Hunenberger 2005; Stoyanov and Groot 2005; Basconi and Shirts 2013).
As noted above, without applying a mechanism to control temperature, the simulations proceed in the microcanonical ensemble (NVE) according to Newton’s equation of motion:
in which m, r and F are the mass, position and force acting on each atom i. The instantaneous temperature of the system (T) is directly related to the particle velocities via,
where k is the Boltzmann constant, N is the number of atoms each of which has mass m and velocity v. Thus, to control the temperature either the velocities of the particles or the equation of motion itself is modified. All of these approaches have the effect of coupling the system to a fictitious heat bath, and the degree of coupling determines how quickly deviations in from the target temperature are reined in. For understanding the influence of the thermostat algorithm on dynamic properties, it is useful to divide them into two categories following the lead of Basconi and Shirts (2013).
In the first group of algorithms, the velocities of particles are rescaled at regular intervals to bring the average temperature back towards the target value. This can be done by directly scaling the velocity of every particle individually, or more commonly be rescaling them together by a common factor such that the target temperature is automatically achieved. This second approach is the ‘direct rescaling’ method examined below. Alternatively, the rescaling can be done in a more subtle way so that the temperature decays more slowly towards the target value as in the Berendsen thermostat (Berendsen et al. 1984) In this the particle velocities are scaled by a common factor such that the deviation of the instantaneous temperature from the target value (T 0) decays with a time constant τ:
The Nose–Hoover method also scales the temperature so that it relaxes in an oscillatory manner towards the target value, but this is done using an extended system that incorporates terms relating to the heat bath (η) in the equation of motion (Nose 1984; Hoover 1985). Here, the velocities are scaled by a variable \(p_{{\eta}} = Q{\raise0.7ex\hbox{$d \eta$} \!\mathord{\left/ {\vphantom {d {dt}}}\right.\kern-0pt} \!\lower0.7ex\hbox{${dt}$}}\) equivalent to the momentum of the heat bath η of mass Q which is coupled to each particle and the instantaneous temperature of the system via the following coupled equations of motion
In the second group of algorithms, particle velocities are randomised rather than being scaled. In the Andersen thermostat, this is done by randomly choosing new velocities for some of the particles at regular intervals from a Boltzmann distribution at the desired temperature (Andersen 1980). The Lowe–Andersen thermostat used in our study is a modification of this, in which the relative velocity of pairs of particles is randomized instead of the velocities of individual particles themselves (Koopman and Lowe 2006). This has the advantage of conserving linear and angular momentum. In Langevin dynamics, the equation of motion is modified to include additional frictional (γ) and random forces (R) on the individual particles
The choice of the friction constant (which is usually the same for all atoms in the system) and the strength of the random forces will dictate the final system temperature.
3 Results
To assess the magnitude of water flux, whether the water flow through single-walled carbon nanotubes is frictionless, and how this is influenced by the choice of thermostat, we determine the pressure-driven flow in (10,10) armchair-type carbon nanotubes (diameter 1.4 nm) of three different lengths (1.4, 5 and 10 nm) using four different thermostats: a direct velocity rescaling method, Lowe–Andersen, Langevin and a modified Langevin thermostat. Published data from pressure-driven simulations of (8,8) carbon nanotubes under a Nose–Hoover thermostat (Su and Guo 2012) and a (7,7) nanotube with a Berendsen thermostat (Nicholls et al. 2012a) is also shown for comparison. While these studies employ slightly different water–CNT interactions and the later uses a TIP4P water model rather than TIP3P, we can still gain qualitative information about how the flow rate changes with nanotube length. Our simulation system comprises twelve nanotubes placed in a 3 × 4 hexagonal array to form a membrane which is bounded on either side by a water box. A hydrostatic pressure difference is generated across the membrane using a method developed by Zhu and Schulten (2003) and implemented in many studies. In this, a force in the direction of the nanotube axis is applied to the oxygen atoms of water molecules in the half of each solvation box most distant from the membrane, as illustrated in Fig. 1. The water flux through the nanotubes was determined for a range of pressures (~50–400 MPa), allowing us to calculate the permeability of the membrane from the slope of the flux versus pressure graph. Additional simulation details are given in the supplementary material.

An example of the main simulation system used in this study. A 4 × 3 array is made from (10,10) carbon nanotubes which separate two water-filled reservoirs. A periodic system in each dimension is used, and force is applied to water oxygen atoms in the pressure application region to generate a hydrostatic pressure difference across the nanotube array. Studies are made of nanotubes of 3 different lengths: 1.4, 5 and 10 nm
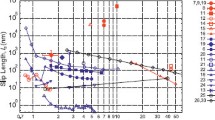
In Fig. 2a, we show how the water flux through the 1.4-nm-long nanotube changes with the hydrostatic pressure difference using four different thermostating methods. In all cases, the flux scales linearly with pressure as has been observed previously (Corry 2008), but the magnitude of the flux differs significantly with thermostat choice. Notably, the flux is almost thrice larger for the direct rescaling and Lowe–Andersen thermostats than it is using the Langevin thermostat with the default-coupling parameters in NAMD. Clearly, the choice of thermostat can influence the calculated water flux or flow enhancement found in simulations.

Simulated water permeabilities with different thermostats. a The water flux at various pressures for a 4 × 3 array of 1.4–nm-long (10,10) carbon nanotubes for four different thermostats. A line of best fit is shown for each thermostat. b The permeability of 4 × 3 array of (10,10) carbon nanotubes of various length using different thermostat methods. c The percentage permeability relative to the shortest nanotube system found with different thermostats. Nanotubes are either constructed as 4 × 3 arrays, or as a single nanotube between a graphene bilayer. Values for the Nose–Hoover and Berendsen thermostats are taken from (Su and Guo 2012) and (Nicholls et al. 2012a), respectively. d The water flux through a 4 × 3 array of 1.4-nm-long, (10,10) carbon nanotubes at 410 MPa for various values of the partial Langevin dynamics-coupling coefficient
Ideally, we would like to compare the flow rates predicted by the simulations reported here with equivalent experimental measurements in order to know which protocol produces the best results. However, such a comparison is far from straightforward for two major reasons. Firstly, there are significant differences in the magnitude of flow rates seen in the various experimental studies (Ritos et al. 2014). The major reason for this is that the experiments are conducted on different membranes containing different types of nanotubes, constructed in different ways. Secondly, the experiments are all made on systems containing a large number of nanotubes whose precise physical characteristics (such as length, width, chirality and defects) are not well defined. In contrast, simulations such as this are typically made on a small number of well-defined pristine nanotubes. While these simulations do see very rough order of magnitude agreement in terms of flow enhancement over macroscopic theory as in experiment, (Ritos et al. 2014) a precise comparison of experimental and simulated flow rates cannot be made until the gulf between the systems being studied is overcome. This study aims to start bridging this gulf by determining inconsistencies in the simulation protocol.
Many simulation studies have suggested that the water flow through carbon nanotubes is near-frictionless, which implies that the permeability of the nanotubes is independent of their length. To assess whether the thermostat algorithm can influence the observed friction in the tubes, in Fig. 2b we plot the water permeability as a function of the nanotube length, while in Fig. 2c we show the permeability as a percentage of that found in the shortest tube. The permeability remains approximately the same across the different tube lengths using direct rescaling and Lowe–Andersen thermostats, implying that there is relatively little friction between water molecules and the interior wall of the nanotube. This conclusion was also reached for the Berensden thermostat (Nicholls et al. 2012a), where it was shown that the flow rate was constant for different lengths of nanotube. The Langevin thermostat, however, shows a significant decrease in permeability for longer tubes. A similar decrease in permeability with tube length is seen in previously published results employing a Nose–Hoover thermostat (Su and Guo 2012). Remarkably, the choice of thermostating algorithm alters the conclusion as to whether the flow of water in nanotubes is frictionless or not.
The Langevin thermostat adds frictional and random forces to the atoms within Newton’s equations of motion, and we expect that both of these could have a significant influence on the simulated water flux in our simulations. It has previously been shown that the randomization of particle velocities inherent in this algorithm can dampen the dynamics of the system, and this is likely to be the cause of the lower flow rate seen with this algorithm compared with the others. To confirm this, we devised an alternative version of the Langevin thermostat which we call the ‘Partial Langevin’ thermostat. In this, frictional and random forces are applied only to water molecules in the upper half of the upper water reservoir and the lower half of the lower reservoir, so that molecules diffusing through the pores are not directly influenced by frictional terms but still maintain temperature via coupling to the reservoirs. As seen in Fig. 2, the permeability is greater using this approach than using the traditional Langevin thermostat, but it is still lower than that found with direct rescaling or the Lowe–Andersen method. In addition, a decrease in permeability with length still occurs in this case, although it is not as marked as for the unmodified Langevin thermostat. While it may be possible to accurately capture transport in the nanotube by coupling to a large enough reservoir, this is not likely to be computationally feasible. Our results clearly show that applying the Langevin thermostat to water molecules inside the nanotubes does influence the simulated flow rate and length dependence of this flow.
As the random force in the Langevin thermostat applies separately to each atom in the system, it can act to reduce any coupled motion of atoms. This is likely to be important in these simulations where water molecules are seen to move in a ‘plug-like’ collective manner through the nanotube (Hummer et al. 2001; Corry 2008; Majumder and Corry 2011; Ritos et al. 2014). As the nanotubes get longer, we expect more atoms to be moving in this collective manner, and so any term which reduces such coupled motion will become more apparent. The Nose–Hoover thermostat similarly adds random forces to atoms which would act to dampen dynamics and remove collective motion which may explain the length dependence of water fluxes seen in that case. In contrast, thermostating algorithms that act globally to adjust the velocities of all atoms (such as velocity rescaling) or that only adjust the relative motion of atom pairs (e.g. Lowe–Andersen) will not act to reduce collective motion in this way, which may explain why no length dependence of flow is seen.
The frictional term in the Langevin equation could also influence the length dependence of flow rates. The size of the frictional term is controlled by the so-called damping coefficient which is typically (although not necessarily) the same for all atoms in the system. The use of small damping coefficients yields poor temperature control, while adding a larger coefficient dissipates kinetic energy more quickly and reduces variations in temperature. However, large frictional or random forces can perturb the dynamics of the system (Hunenberger 2005; Basconi and Shirts 2013). To see how the choice of damping coefficient alters the flux of water through the nanotubes in our simulation system, we calculate the water flux using a variety of damping coefficient values with the partial Langevin algorithm. As can be seen in Fig. 2d, the use of larger frictional terms reduces the net flux through the nanotubes, and the absolute value varies by almost tenfold across the range studied. An even greater influence of damping coefficient on water flux should be expected with the unmodified Langevin algorithm. Damping coefficients in the range of 2–15 ps−1 are typically used in MD simulations (with a default value of 5 ps−1 found in NAMD), and these results show that careful consideration needs to be given when choosing a value if mass transport is being measured. As suspected from previous investigations, (Basconi and Shirts 2013) results with the Langevin thermostat should approach those found in a microcanonical NVE ensemble (no temperature control) in the limit of weak damping.
4 Discussion
That the Langevin algorithm alters the dynamics of our pressure-driven system is not unexpected. Indeed, as has been noted in previous studies, the Langevin thermostat is not intended to be used to determine mass transport properties; instead it focuses on replicating energetics and maintaining dynamic stability (Hunenberger 2005). However, given that this thermostat and the specific choice of coupling parameters can alter the simulated permeability of the nanotubes by several orders of magnitude and that they can influence the dependence of permeability on tube length provides a note of caution in both quantitative and qualitative studies.
As has been discussed elsewhere, the choice of an appropriate thermostat for specific MD applications is a subjective matter, with each approach having advantages and disadvantages (Hunenberger 2005; Stoyanov and Groot 2005; Basconi and Shirts 2013). The Langevin thermostat is dynamically stable and reproduces a canonical ensemble over long timescales, but the frictional and random forces can perturb the mass transport properties. While this can be minimised by only controlling the temperature in the distant reservoirs as in our partial Langevin method, extreme care must be taken in the choice of atomic damping coefficients. A decrease in nanotube permeability with nanotube length is also observed with the Nose–Hoover thermostat, (Su and Guo 2012) producing similar results to those found with the Langevin thermostat. In contrast, results with direct rescaling, Berendsen and Lowe–Andersen thermostats both show no reduction in flow rate with nanotube length, although it is known that the Berendsen thermostat will not always reproduce the correct energy distribution. These results support previous findings that algorithms that randomise particle velocities (e.g. Langevin, Andersen) dampen the dynamics of the system, while those that effectively scale particle velocities (e.g. Berendsen, direct rescaling) do not (Basconi and Shirts 2013). The Nose–Hoover thermostat does give a good depiction of dynamics in equilibrium simulations, but performs less well here, presumably due to a damping of coupled motion. The Nose–Hoover thermostat is also not Galilean invariant, meaning that centre of mass motion such as seen in these simulations of mass transport has to be corrected for, otherwise it is seen as an increase in system temperature (Koopman and Lowe 2006).
Dissipative particle dynamics (DPD) is another approach for controlling molecular simulation (Groot and Warren 1997) that could avoid some of the problems experienced by the Langevin thermostat as it conserves momentum. While it also includes frictional and random terms, these forces are applied to particle pairs rather than individual particles, avoiding the damping of coupled motion. Alternatively, altering velocity only perpendicular to the flow would prevent the damping of the transport rate, although doing this using a velocity randomisation scheme may still dampen local motion.
Although not considered here, other simulation parameters such as the choice of water model could also influence simulated flow rates. For example, the self-diffusion coefficients found with different water models differ by up to threefold from each other and from experimental values (Mark and Nilsson 2001) which may also alter the rate of pressure-driven flow rates.
5 Conclusions
By conducting simulations of water transport in carbon nanotubes under a hydrostatic pressure difference with a variety of thermostats, we are able to show that the means of controlling simulation temperature can have profound implications on the observed transport properties. This knowledge helps to reconcile disparate simulation results which have predicted quantitative water permeabilities that differ by over an order of magnitude, as well as qualitative differences in the effect of changing the nanotube length. Furthermore, it provides a warning that the selection of which thermostat to employ and to which atoms it is applied should be carefully considered when conducting simulations of mass transport. These results suggest that existing studies using the Langevin or Nose–Hoover thermostats may be underestimating the likely nanotube permeability and may overestimate the reduction in flow with increasing nanotube length. These results support the claims of extremely and near-frictionless water flow in carbon nanotubes.
References
Andersen HC (1980) Molecular-dynamics simulations at constant pressure and-or temperature. J Chem Phys 72:2384–2393
Basconi JE, Shirts MR (2013) Effects of temperature control algorithms on transport properties and kinetics in molecular dynamics simulations. J Chem Theory Comput 9:2887–2899
Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR (1984) Molecular-dynamics with coupling to an external bath. J Chem Phys 81:3684–3690
Cohen-Tanugi D, Grossman JC (2012) Water desalination across nanoporous graphene. Nano Lett 12:3602–3608
Corry B (2008) Designing carbon nanotube membranes for efficient water desalination. J Phys Chem B 112:1427–1434
Corry B (2011) Water and ion transport through functionalised carbon nanotubes: implications for desalination technology. Energ Environ Sci 4:751–759
Gong XJ, Li JY, Zhang H, Wan RZ, Lu HJ, Wang S, Fang HP (2008) Enhancement of water permeation across a nanochannel by the structure outside the channel. Phys Rev Lett 101:257801
Gong XJ, Li JC, Xu K, Wang JF, Yang H (2010) A controllable molecular Sieve for Na+ and K+ ions. J Am Chem Soc 132:1873–1877
Groot RD, Warren PB (1997) Dissipative particle dynamics: bridging the gap between atomistic and mesoscopic simulation. J Chem Phys 107:4423–4435
Hilder TA, Gordon D, Chung SH (2009) Salt rejection and water transport through boron nitride nanotubes. Small 5:2183–2190
Hilder TA, Yang R, Ganesh V, Gordon D, Bliznyuk A, Rendell AP, Chung SH (2010) Validity of current force fields for simulations on boron nitride nanotubes. Micro Nano Lett 5:150–156
Holt JK, Park HG, Wang YM, Stadermann M, Artyukhin AB, Grigoropoulos CP, Noy A, Bakajin O (2006) Fast mass transport through sub-2-nanometer carbon nanotubes. Science 312:1034–1037
Hoover WG (1985) Canonical dynamics—equilibrium phase-space distributions. Phys Rev A 31:1695–1697
Hummer G, Rasaiah JC, Noworyta JP (2001) Water conduction through the hydrophobic channel of a carbon nanotube. Nature 414:188–190
Hunenberger P (2005) Thermostat algorithms for molecular dynamics simulations. Adv Polym Sci 173:105–147
Joseph S, Aluru NR (2008) Why are carbon nanotubes fast transporters of water? Nano Lett 8:452–458
Kalra A, Garde S, Hummer G (2003) Osmotic water transport through carbon nanotube membranes. P Natl Acad Sci USA 100:10175–10180
Kannam SK, Todd BD, Hansen JS, Daivis PJ (2013) How fast does water flow in carbon nanotubes? J Chem Phys 138:094701
Khademi M, Sahimi M (2011) Molecular dynamics simulation of pressure-driven water flow in silicon-carbide nanotubes. J Chem Phys 135:204509
Koopman EA, Lowe CP (2006) Advantages of a Lowe–Andersen thermostat in molecular dynamics simulations. J Chem Phys 124:204103
Krishnan TVS, Babu JS, Sathian SP (2013) A molecular dynamics study on the effect of thermostat selection on the physical behaviour of water molecules inside single walled carbon nanotubes. J Mol Liq 188:42–48
Li PH, Lim XD, Zhu YW, Yu T, Ong CK, Shen ZX, Wee ATS, Sow CH (2007) Tailoring wettability change on aligned and patterned carbon nanotube films for selective assembly. J Phys Chem B 111:1672–1678
Liu HM, Murad S, Jameson CJ (2006) Ion permeation dynamics in carbon nanotubes. J Chem Phys 125:084713
Majumder M, Corry B (2011) Anomalous decline of water transport in covalently modified carbon nanotube membranes. Chem Commun 47:7683–7685
Majumder M, Chopra N, Andrews R, Hinds BJ (2005) Nanoscale hydrodynamics: enhanced flow in carbon nanotubes (vol 438, p 44, 2005). Nature 438:930
Mark P, Nilsson L (2001) Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A 105:9954–9960
Nicholls WD, Borg MK, Lockerby DA, Reese JM (2012a) Water transport through (7,7) carbon nanotubes of different lengths using molecular dynamics. Microfluid Nanofluid 12:257–264
Nicholls WD, Borg MK, Lockerby DA, Reese JM (2012b) Water transport through carbon nanotubes with defects. Mol Simulat 38:781–785
Nose S (1984) A unified formulation of the constant temperature molecular-dynamics methods. J Chem Phys 81:511–519
Peter C, Hummer G (2005) Ion transport through membrane-spanning nanopores studied by molecular dynamics simulations and continuum electrostatics calculations. Biophys J 89:2222–2234
Ritos K, Mattia D, Calabro F, Reese JM (2014) Flow enhancement in nanotubes of different materials and lengths. J Chem Phys 140:014702
Rosta E, Buchete NV, Hummer G (2009) Thermostat artifacts in replica exchange molecular dynamics simulations. J Chem Theory Comput 5:1393–1399
Song C, Corry B (2009) Intrinsic ion selectivity of narrow hydrophobic pores. J Phys Chem B 113:7642–7649
Stoyanov SD, Groot RD (2005) From molecular dynamics to hydrodynamics: a novel Galilean invariant thermostat. J Chem Phys 122:114112
Su JY, Guo HX (2012) Effect of nanochannel dimension on the transport of water molecules. J Phys Chem B 116:5925–5932
Suk ME, Raghunathan AV, Aluru NR (2008) Fast reverse osmosis using boron nitride and carbon nanotubes. Appl Phys Lett 92:133120
Thomas JA, McGaughey AJH (2008) Reassessing fast water transport through carbon nanotubes. Nano Lett 8:2788–2793
Thomas JA, McGaughey AJH, Kuter-Arnebeck O (2010) Pressure-driven water flow through carbon nanotubes: insights from molecular dynamics simulation. Int J Therm Sci 49:281–289
Won CY, Aluru NR (2007) Water permeation through a subnanometer boron nitride nanotube. J Am Chem Soc 129:2748–2749
Won CY, Aluru NR (2008) Structure and dynamics of water confined in a boron nitride nanotube. J Phys Chem C 112:1812–1818
Won CY, Joseph S, Aluru NR (2006) Effect of quantum partial charges on the structure and dynamics of water in single-walled carbon nanotubes. J Chem Phys 125:114701
Zhu FQ, Schulten K (2003) Water and proton conduction through carbon nanotubes as models for biological channels. Biophys J 85:236–244
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Thomas, M., Corry, B. Thermostat choice significantly influences water flow rates in molecular dynamics studies of carbon nanotubes. Microfluid Nanofluid 18, 41–47 (2015). https://doi.org/10.1007/s10404-014-1406-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-014-1406-y