
Abstract
Integrated PCR–CE chip technology has immense potential to be applied in clinical diagnostics. In this work we demonstrate the application of our integrated PCR–CE chip for the detection of the respiratory pathogen Bordetella pertussis. A series of experiments with varying cell concentrations (200,000–2 cfu) were performed to obtain the analytical detection limits of the chip. We find that the chip technology is well suited for sensitive detection of Bordetella pertussis, using genetic material from less than even 2 cfu. We also utilized an off-chip real-time PCR method to compare and validate our on-chip approach.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The microfluidic integration of polymerase chain reaction (PCR) (Kricka and Wilding 2003) with capillary electrophoresis (CE) (Harrison et al. 1992; Manz et al. 1993) has emerged as a promising chip based approach for rapid and low-cost genetic analysis. Using different integration strategies, PCR–CE devices have recently been demonstrated as a useful tool for many clinical and forensic applications (Bienvenue et al. 2006; Easley et al. 2006; Ferrance et al. 2005; Koh et al. 2003; Lagally et al. 2001; Li and Kricka 2006; Lin et al. 2000; Liu et al. 2007; Matsunaga et al. 2003; Rodriguez et al. 2003; Sethu and Mastrangelo 2004; Shandrick et al. 2002; Wang et al. 2004; Waters et al. 1998).
More recently, pathogen disease detection using chip based PCR–CE has become a topic of extreme interest, because of the growing number of outbreaks. With the recent outbreaks of severe acute respiratory syndrome (SARS), E. coli, influenza, anthrax, to name a few, it is important for the microfluidic research community to leverage the capabilities of chip based detection technologies. In this regard, many microfluidic research groups have made significant contribution (Easley et al. 2006; Lagally et al. 2004; Liao et al. 2005; Pal et al. 2005; Zhou et al. 2004), and these demonstrations have a good potential of being capitalized by healthcare professionals in a clinical setting.
Another highly contagious pathogen is Bordetella pertussis (B. pertussis), the symptoms, of which are commonly referred to as “whooping cough” (Knorr et al. 2006; Mattoo and Cherry 2005). B. pertussis is a small, gram-negative rod shaped pathogen that infects the respiratory tract, with the complication of pulmonary hypertension, which can prove fatal (Casano et al. 2002). Humans are the only known hosts for this pathogen, with infants and adolescents being the most susceptible (Hewlett and Edwards 2005; Weir 2002). In 2004 more than 25,000 cases of B. pertussis were reported, a 12-fold increase since 1980 (Hewlett and Edwards 2005). Thus, a means to rapidly detect B. pertussis and facilitate large scale screening for outbreaks is becoming increasingly important.
A number of recent independent studies have concluded that PCR amplification and detection methods for this pathogen is superior to available traditional detection methods and should be routinely incorporated in clinical testing for screening and surveillance (Massay 2007; Muller et al. 1997; Tatti et al. 2006). Importantly, since PCR only requires a small quantity of genetic material (i.e., capable of amplifying even a single DNA strand) it facilities the early detection of B. pertussis, thereby greatly improving targeted remedial medical care with improved outcomes (Davis 2005; Tatti et al. 2006). In addition to the advantages of conventional PCR based detection of B. pertussis, integrated PCR–CE chip technology provides a number of added benefits. These include: low reagent consumption, minimal cross-contamination, and a potentially cost-effective detection strategy leading to routine nucleic acid testing/screening.
Herein we apply a previously developed integrated PCR–CE chip (Prakash and Kaler 2007) for on-chip genetic amplification (by PCR) and subsequent detection (utilizing CE) of the B. pertussis respiratory pathogen. Furthermore, we determined the analytical detection limits of the PCR–CE chip by performing on-chip experiments with various known concentration of B. pertussis. The on-chip data was also compared with an off-chip laboratory “gold standard”, real-time PCR, to facilitate validation and reliability of the chip based approach. We conclude that this PCR–CE chip based technology can readily detect B. pertussis from genetic material equivalent to about the content of a cell.
2 Experimental methods
2.1 Integrated PCR–CE chip
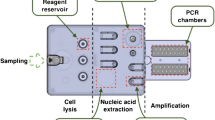
The integrated PCR–CE chip, as reported in prior work (Prakash and Kaler 2007), is shown in Fig. 1. It comprises of an array of nine PCR sample chambers with associated valved fluidic ports (input/output) on either side of each chamber. These chambers are in turn connected via fluidic channels to the CE section of the chip. The CE separation uses a sieving medium (Genescan polymer, Applied Biosystems, USA) and TBE buffer protocol. The high voltage for CE and optical detection system for laser induced fluorescence (LIF) is housed in the μTk (Micralyne, Canada). A detailed description of the chip, operating principles and utility for genetic analysis can be found elsewhere (Prakash and Kaler 2007).

A schematic of the integrated PCR–CE chip with the different labelled components. The PCR section has nine independent chambers for simultaneous PCR amplification. These chambers are in turn connected to the CE section by an interconnecting channel for subsequent sequential CE analysis
2.2 B. pertussis culture and estimation of concentration
B. pertussis was grown in standard cultures at 35°C in an ambient atmosphere on Regan-Lowe media (Daylnn Biologicals, Canada), as described previously (Knorr et al. 2006). The cells were typically cultured for 2 days prior to experimentation. A small sample (20 μl) of B. pertussis culture was suspended in sterile PCR grade water (Fluka Biochemika, Switzerland) at a concentration equivalent to 1 × 107 colony forming units (cfu)/ml based on the McFarland turbidimetric standard (Knorr et al. 2006) (Note: 1 cfu/ml refers to 1 live B. pertussis cell/ml). A serial dilution of this 1 × 107 cfu/ml B. pertussis preparation was then performed in order to achieve aliquots of lower concentration.
Table 1 gives the B. pertussis cell concentration of the stock culture and the final cell concentration of the serial dilutions (1 × 107–1 × 101 cfu/ml, labelled as A7–A1). DNA was independently extracted from each of these samples and suspended in 50 μl of PCR-grade water. These samples were then utilized in a series of on-chip PCR and real-time PCR experiments, as described in the following section.
2.3 PCR assay
The PCR primers were specifically designed to amplify a 167 bp fragment in the IS481 region for detection of B. pertussis, as previously described (Knorr et al. 2006) and adopted from Reischl et al. (Reischl et al. 2001). Since B. pertussis carries over 50 copies of the IS481, it is a desirable target for sensitive detection of this pathogen (Knorr et al. 2006). The primer sequences were: (forward) 5′-GCGTGCAGATTCGTCGTAC3′ and (reverse) 5′TGATGGTGCCTATTTTACGG3′. For on-chip PCR–CE, the forward primer was labelled with the fluorophore VIC, while for the real-time PCR a (TaqMan) hybridization probe (5′FAM-ACCCTCGATTCTTCCGT-BNFQ3′) was used. The primers and probes were synthesized and purchased from Applied Biosystems, USA. All PCR reagents were purchased from Invitrogen (Canada) and the assays for both on-chip and off-chip real-time PCR were similar, with final component concentrations as follows: 1x PCR buffer, 0.2 mM each dNTP, 2 mM MgCl2, 2.5U Taq, 0.5 mM each primer, and PCR grade water (Fluka Biochemika, Switzerland). The assay involved an initial denaturing at 95°C for 10 min, followed by 40 cycles of denaturing (95°C), annealing (50°C), extension (72°C) of 15 s each, and a final extension (72°C) for 10 min.
For PCR performed on-chip, 1 μl of the extracted DNA was included in 15 μl of PCR master mix. However, only ∼3 μl of the PCR master-mix was loaded into the PCR chamber. This large volume of PCR master-mix was primarily prepared to minimize pipetting errors and facilitate multiple on-chip runs. Off-chip real-time PCR assay for a ‘‘gold-standard’’ comparison was prepared separately with 5 μl of the suspended DNA in 15 μl of PCR master mix. Real-time PCR was performed using the LightCycler® instrument (Roche Diagnostics, Canada). All on-chip PCR and off-chip real-time PCR experiments were performed simultaneously and under identical assay preparation conditions to facilitate a comparison of the data. Negative PCR controls (i.e., PCR assay without the addition of DNA) were also run for both on-chip and real-time to ensure that cross and carry-over contaminations were avoided.
Upon completion of PCR thermal cycling on the integrated chip, a portion of the PCR sample was subsequently utilized for the on-chip CE analysis of the PCR amplified DNA using the CE protocol as described in earlier work (Prakash and Kaler 2007). The on-chip CE analysis required only 0.3 μl of the amplified PCR sample, i.e., about one-tenth of the PCR volume contained in the chamber. In the case of the real-time PCR, single fluorescent readings were taken at the annealing temperature 55°, once per cycle. Amplification, detection and data analysis for real-time PCR were conducted utilizing the LightCycler® 1.0 software version 4.05 (Roche Diagnostics, Canada).
3 Results: PCR assay
A plot of relative fluorescence units (RFU) verses time (i.e., CE electropherogram), for the B. pertussis PCR amplified DNA utilizing the integrated PCR–CE chip is shown in Fig. 2. Chip based PCR was performed using sample A3 (see Table 1). As seen in Fig. 2, a strong primer peak is observed at 190 s and a product peak (i.e., B. pertussis amplified DNA) is seen at ∼212 s, confirming the successful amplification of the IS481 region. To verify the size of this resulting PCR product, we included size standards (Gene Scan 500, Applied Biosystems, USA) in this CE separation analysis. Hence a number of smaller peaks corresponding to different fragment sizes are also observed in the CE separation, Fig. 2. These standards confirmed that the B. pertussis amplified PCR product is 167 bp, as designed, positioned between fragment sizes 160 bp (210 s) and 200 bp (216 s).

CE analysis [relative fluorescence intensity (RFU) vs. time in s] of B. pertussis PCR amplification on the integrated chip. The 167 bp PCR product (DNA) is observed at 212 s and the molecular size was verified by separation with Gene Scan 500 size standards
In another set of experiments (see Table 1), PCR followed by CE analysis of samples A1–A4 and A7 were performed using the integrated chip, the CE analysis, of which is shown in Fig. 3a. The CE data shows very strong PCR products for the three samples A3, A4 and A7, for which the DNA was extracted from cell concentrations above 20 cfu. As observed in Fig. 3a, the fluorescence signal reached a maximum value of 5 RFU for these samples, which indicates optimal amplification and an easily identifiable species. Next, for sample A2, DNA extracted from only 2 cfu, a weak (although distinctly detectable) PCR product peak is observed, with a fluorescence signal of ∼0.5 RFU. Finally, for sample A1, since the theoretically estimated cell concentration was a fraction of a single cell, no detectable amplification signal was present indicative of the absence of the target DNA in the sample. [Note: Analysis of sample A5 (2000 cfu) and A6 (2 × 104 cfu) is not presented here since all samples above 20 cfu (i.e., A3–A7) provide very high PCR amplification with CE/LIF fluorescence maxed at 5 RFU.]

a The 3D plot shows the CE analysis of different concentrations of B. pertussis samples run on the integrated chip. Samples A7, A4 and A3 had a maximum RFU of 5, sample A2 had a RFU of 0.5, while A1 showed no fluorescence signal. For clarity, the PCR product “Time vs RFU” for samples A2 and A1 are shown in magnified inserts. b Real-time PCR LightCycler® amplification profile of the IS481 hybridization probe assay with a dilution series of B. pertussis, for samples A7–A1. In CE the PCR amplification is analysed based on the fluorescence intensity of the amplified DNA and time, while in real-time PCR it is analysed at the amplification stage itself by the fluorescence of the hybridization probe at each thermal cycle
In an identical set of experiments with samples A1–A7 using real-time PCR for “gold–standard” comparison, we note distinct amplification signals for each sample A2–A7, as shown in Fig. 3b. Since the PCR amplification data is obtained after each thermal cycle, a unique crossing point (CP) is also observed for each sample, as indicated in Fig. 3b and summarized in Table 1. CP is the cycle number, at which the amplification curve becomes exponential. A high CP value indicates low initial copy number of IS481 amplification targets in a sample and vice versa. Hence, as summarized in Table 1, sample A7 has a CP value of 14.86 while A2 only has a CP of 34.98. This data, as shown by Knorr et al. (2006) and others, provides easy extrapolation of information that can be correlated with the quantity of cells in the initial sample.
Negative control PCR assays for both on-chip and off-chip methodologies, did not indicate any detectable amplification (data not shown), suggesting a lack of contamination by careful assay preparation. All experiments presented here we repeated at least thrice more than the data presented herein and yielded similar results. While stochastic amplification effects were not noted in the current study, we anticipate that for clinical field tests involving large sample numbers, stochastic effects may be observed at low genetic concentrations (e.g., sample A2).
4 Discussion
The on-chip PCR–CE analysis, as described, showed easily detectable signal for the identification of B. pertussis from DNA extracted from varying cell concentrations, as low as 2 cfu to as high as 2 × 105 cfu (samples A2–A7). Of these, the successful identification of B. pertussis using sample A2 is of particular interest as it describes the extremely low analytical detection capabilities of the on-chip approach. Sample A2, which consisted of only 2 cfu, the extracted DNA, of which was suspended in 50 μl and only 1 μl of this suspended DNA was utilized for the on-chip PCR assay, showed on-chip CE/LIF detectable amplification of 0.5 RFU. Theoretically, this implies that a sample with possibly only a fraction of cell content is sufficient to identify the presence of B. pertussis. While this theoretically derived fractional content may appear to be inapplicable, it may in fact be critical when applied in routine clinical testing. For example, the presence of even a single B. pertussis cell may well be sufficient to definitively detect the presence of the pathogen. Furthermore, in real-life worst case scenarios, this very low (≤2 cfu) on-chip analytical detection capabilities can easily accommodate potential loss of cell genetic material during handing, limitations with DNA isolation, on-chip inefficiency due to increased surface-to-volume ratios, or other commonly encountered intricacies with chip-based approaches (Prakash et al. 2006, 2007).
Another aspect of the work presented herein was the comparison of chip based B. pertussis identification with the ‘‘gold-standard’’ real-time PCR method. As evident from a direct comparison, real-time PCR provided details on the exact thermal cycle, at which the DNA amplification begins to occur and the possibility to re-trace the cell concentration. This perhaps is particularly useful in a research laboratory environment for the development of an optimized PCR assay protocol (e.g., titrate the many reagents, DNA-primer melting curve determination, etc.) and for identifying the disease state of the patient based on cell count. However, such details are not readily extrapolated from a chip based CE electropherogram, since the method provides data upon completion of the PCR. Nevertheless, CE chip based analysis is being extensively promoted for future clinical analysis devices (Li and Kricka 2006). This is primarily because chip based approaches are targeted for integrated and automated, miniaturized, low-cost, portable point-of-care devices that may not require detailed data extrapolation for assay optimization. Since the chip based approach is comparable to off-chip methods in terms of analytical detection limits, the ability of the chip method to provide an assertive binary data (i.e., presence or absence) of a specimen in a sample would serve the purpose of improved and early detection very well. For example, samples identified as positive by low-cost chip detection technology, could then be referred for a more detailed laboratory analysis to facilitate tailored therapy. Clearly, the chip technology would cater to many critical healthcare requirements such as low-cost, mass and routine screening leading to early detection of infection and outbreaks, ultimately leading to better cost-effective healthcare management.
For the detection of B. pertussis, the presence of cell debris in the PCR assay adversely affects the efficiency of the amplification to varied extents, sometimes causing complete inhibition (Loeffelholz et al. 1999). Hence to avoid such variables, herein we utilized purified B. pertussis extracted DNA to determine the detection range/limits of the chip. In future work we anticipate incorporation of on-chip lysis with purified DNA extraction to facilitate more rapid detection.
In conclusion, in this work we demonstrated an integrated PCR–CE chip method for the identification of B. pertussis. This PCR–CE chip used <3 μl for PCR, of which only 0.3 μl was utilized for subsequent CE analysis on the integrated chip. Conceivably the PCR could be performed with one-tenth the current volume, sufficient for detection of low copy number B. pertussis, which will further reduce reagent costs, greatly surpassing the limitations of conventional off-chip methods. Furthermore in this work, a set of B. pertussis cell concentration serial dilution experiments confirmed that the integrated chip is capable of identifying B. pertussis from the genetic content of ≤2 cells. Currently efforts are in progress to integrate upstream sample preparation such as cell pre-concentration using dielectrophoresis with the herein demonstrated downstream PCR–CE detection.
Clearly, the advantage of the chip technology is to harness low-cost screening for pathogen/infectious disease identification. It is anticipated that costs of such a procedure would be reduced promoting improved healthcare management strategies to prevent disease outbreaks.
References
Bienvenue JM, Duncalf N, Marchiarullo D, Ferrance JP, Landers JP (2006) Microchip-based cell lysis and DNA extraction from sperm cells for application to forensic analysis. J Forensic Sci 51(2):266–273
Casano P, Odena MP, Cambra FJ, Martin JM, Palomeque A (2002) Bordetella pertussis infection causing pulmonary hypertension. Arch Dis Child 86(6):453–454
Davis JP (June 2005) Clinical and economic effects of pertussis outbreaks. Pediatr Infect Dis J 24(6)
Easley CJ, Karlinsey JM, Bienvenue JM, Legendre LA, Roper MG, Feldman SH, Hughes MA, Hewlett EL, Merkel TJ, Ferrance JP, Landers JP (2006) A fully integrated microfluidic genetic analysis system with sample-in-answer-out capability. Proc Natl Acad Sci USA 103(51):19272–19277
Ferrance JP, Bienvenue J, Legendre L, Easley C, Karlinsey J, Roper MG, Landers JP (2005) A fully integrated microdevice for clinical analysis. Paper presented at the ninth international conference on miniaturized systems for chemistry and life Sciences, MicroTAS (μTAS). Boston, USA
Harrison DJ, Manz A, Fan ZH, Ludi H, Widmer HM (1992) Capillary electrophoresis and sample injection systems integrated on a planar glass chip. Anal Chem 64(17):1926–1932
Hewlett EL, Edwards KM (2005) Pertussis––not just for kids. N Engl J Med 352(12):1215–1222
Knorr L, Fox JD, Tilley PAG, Ahmed-Bentley J (2006) Evaluation of real-time PCR for diagnosis of Bordetella pertussis infection. Bmc Infect Dis 6
Koh CG, Tan W, Zhao MQ, Ricco AJ, Fan ZH (2003) Integrating polymerase chain reaction, valving, and electrophoresis in a plastic device for bacterial detection. Anal Chem 75(17):4591–4598
Kricka LJ, Wilding P (2003) Microchip PCR. Anal Bioanal Chem 377(5):820–825
Lagally ET, Emrich CA, Mathies RA (2001) Fully integrated PCR-capillary electrophoresis microsystem for DNA analysis. Lab Chip 1(2):102–107
Lagally ET, Scherer JR, Blazej RG, Toriello NM, Diep BA, Ramchandani M, Sensabaugh GF, Riley LW, Mathies RA (2004) Integrated portable genetic analysis microsystem for pathogen/infectious disease detection. Anal Chem 76(11):3162–3170
Li SFY, Kricka LJ (2006) Clinical analysis by microchip capillary electrophoresis. Clin Chem 52(1):37–45
Liao CS, Lee GB, Wu JJ, Chang CC, Hsieh TM, Huang FC, Luo CH (2005) Micromachined polymerase chain reaction system for multiple DNA amplification of upper respiratory tract infectious diseases. Biosen Bioelectron 20(7):1341–1348
Lin YC, Huang MY, Young KC, Chang TT, Wu CY (2000) A rapid micro-polymerase chain reaction system for hepatitis C virus amplification. Sens Actuators B Chem 71(1–2):2–8
Liu P, Seo TS, Beyor N, Shin KJ, Scherer JR, Mathies RA (2007) Integrated portable polymerase chain reaction-capillary electrophoresis microsystem for rapid forensic short tandem repeat typing. Anal Chem 79(5):1881–1889
Loeffelholz MJ, Thompson CJ, Long KS, Gilchrist MJR (1999) Comparison of PCR, culture, and direct fluorescent-antibody testing for detection of Bordetella pertussis. J Clin Microbiol 37(9):2872–2876
Manz A, Harrison DJ, Verpoorte E, Widmer HM (1993) Planar chips technology for miniaturization of separation systems––a developing perspective in chemical monitoring. Adv Chromatogr 33:1–66
Massay SC (2007) A survey of state public health laboratories for pertussis diagnostics. Lab Med 38(3):169–171
Matsunaga H, Anazawa T, Yeung ES (2003) Integrated on-capillary instrumentation for gene expression measurement directly from cells. Electrophoresis 24(3):458–465
Mattoo S, Cherry JD (2005) Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin Microbiol Rev 18(2):326–82
Muller FMC, Hoppe JE, vonKonig CHW (1997) Laboratory diagnosis of pertussis: state of the art in 1997. J Clin Microbiol 35(10):2435–2443
Pal R, Yang M, Lin R, Johnson BN, Srivastava N, Razzacki SZ, Chomistek KJ, Heldsinger DC, Haque RM, Ugaz VM, Thwar PK, Chen Z, Alfano K, Yim MB, Krishnan M, Fuller AO, Larson RG, Burke DT, Burns MA (2005) An integrated microfluidic device for influenza and other genetic analyses. Lab Chip 5(10):1024–1032
Prakash R, Kaler KVIS (2007) An integrated genetic analysis microfluidic platform with valves and a PCR chip reusability method to avoid contamination. Microfluidics Nanofluidics 3(2):177–187
Prakash AR, Adamia S, Sieben V, Pilarski P, Pilarski LM, Backhouse CJ (2006) Small volume PCR in PDMS biochips with integrated fluid control and vapour barrier. Sens Actuators B Chem 113(1):398–409
Prakash RA, Amrein M, Kaler KVIS (2007) Characteristics and impact of Taq enzyme adsorption on surfaces in microfluidic devices. Microfluidics Nanofluidics (in press), doi:10.1007/s10404–007–0174–3
Reischl U, L. ehn N, S anden G, L. oeffelholz M (2001) Real-time PCR assay targeting IS481 of Bordetella pertussis and molecular basis for detecting Bordetella holmesii. J Clin Microbiol 39(5):1963–1966
Rodriguez I, Lesaicherre M, Tie Y, Zou QB, Yu C, Singh J, Meng LT, Uppili S, Li SFY, Gopalakrishnakone P, Selvanayagam ZE (2003) Practical integration of polymerase chain reaction amplification and electrophoretic analysis in microfluidic devices for genetic analysis. Electrophoresis 24(1–2):172–178
Sethu P, Mastrangelo CH (2004) Cast epoxy-based microfluidic systems and their application in biotechnology. Sens Actuators B Chem 98(2–3):337–346
Shandrick S, Ronai Z, Guttman A (2002) Rapid microwell polymerase chain reaction with subsequent ultrathin-layer gel electrophoresis of DNA. Electrophoresis 23(4):591–595
Tatti KM, Wu KH, Sanden GN, Greer P, Sumner J, Guarner J, Paddock CD, Zaki SR (2006) Molecular diagnosis of Bordetella pertussis infection by evaluation of formalin-fixed tissue specimens. J Clin Microbiol 44(3):1074–1076
Wang SH, Wen JK, Zhou YF, Zhang ZP, Yang RF, Zhang JB, Chen J, Zhang XE (2004) Identification and characterization of Bacillus anthracis by multiplex PCR on DNA chip. Biosens Bioelectron 20(4):807–813
Waters LC, Jacobson SC, Kroutchinina N, Khandurina J, Foote RS, Ramsey JM (1998) Microchip device for cell lysis, multiplex PCR amplification, and electrophoretic sizing. Anal Chem 70(1):158–162
Weir E (2002) Resurgence of Bordetella pertussis infection. Can Med Assoc J 167(10)
Zhou ZM, Liu DY, Zhong RT, Dai ZP, Wu DP, Wang H, Du YG, Xia ZN, Zhang LP, Mei XD, Lin BC (2004) Determination of SARS-coronavirus by a microfluidic chip system. Electrophoresis 25(17):3032–3039
Acknowledgments
We are grateful for the financial support from, Canadian Institutes of Health Research (CIHR), Natural Science and Engineering Research Council of Canada (NSERC) and Calgary Provincial Laboratory (Alberta, Canada).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Prakash, A.R., De la Rosa, C., Fox, J.D. et al. Identification of respiratory pathogen Bordetella Pertussis using integrated microfluidic chip technology. Microfluid Nanofluid 4, 451–456 (2008). https://doi.org/10.1007/s10404-007-0195-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-007-0195-y