
Abstract
Immunoglobulin G4 (IgG4)-related disease is a novel clinical entity characterized by infiltration of IgG4-immunopositive plasmacytes and elevated serum IgG4 concentration accompanied by enlargement of and masses in various organs, including the lacrimal gland, salivary gland, and pancreas. Recent studies have clarified that conditions previously diagnosed as Mikulicz disease as well as various types of lymphoplasmacytic infiltrative disorders of the ocular adnexa are consistent with a diagnosis of IgG4-related disease. Against this background, the diagnostic criteria for IgG4-related ophthalmic disease have recently been established, based on both the clinical and the histopathologic features of the ocular lesions. This article reviews these new criteria with reference to the comprehensive diagnostic criteria for IgG4-related disease for all systemic conditions reported in 2012.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunoglobulin G4 (IgG4)-related disease is a collection of disorders of unknown etiology that are characterized by an infiltrate of IgG4-immunopositive plasmacytes accompanied by enlargement of and masses, nodules, or hypertrophic lesions in various organs of the body. The comprehensive diagnostic criteria for IgG4-related disease were reported in 2012 by the Research Program for Intractable Disease of the Ministry of Health, Labor, and Welfare (MHLW) Japan, All Japan IgG4 team (Table 1) [1]. The disorder, which used to be diagnosed as Mikulicz disease, involves symmetric enlargement of the bilateral lacrimal and salivary glands. Recent studies have found that most cases of Mikulicz disease are consistent with a diagnosis of IgG4-related disease. With this finding, ocular lesions related to IgG4 have attracted attention. However, in addition to the fact that the disorder is a relatively new disease entity, the various disease names used in the past to describe IgG4-related ocular lesions and the lack of definitive diagnostic criteria have created some confusion in the clinical setting as well as among professional bodies.
To address this situation, the ophthalmology committee composed of members of the Japanese Study Group for IgG4-Related Ophthalmic Disease in the Research Program for Intractable Disease (IgG4-related disease) of the Japanese Ministry of Health, Labor, and Welfare has taken the lead in developing diagnostic criteria for IgG4-related ophthalmic disease. This article reports on the background and criteria proposed by the group.
History of IgG4-related disease
IgG, one of the immunoglobulins in serum, is composed of four subclasses: IgG1–IgG4. Among these subclasses, IgG4 constitutes only approximately 4 % of the total IgG. In 2001, Hamano et al. [2] reported abnormally high serum IgG4 levels in patients with autoimmune pancreatitis. Subsequent to their report, the association of IgG4 with various systemic diseases began to unfold. Apart from pancreatitis, cases showing enlargement or hypertrophy of various organs, such as the lacrimal gland (Fig. 1), salivary gland, hepatobiliary tract, and retroperitoneum, have been found to have elevated serum IgG4 levels. Subsequent studies gradually identified more characteristics of this group of diseases, such as marked infiltration of IgG4-positive plasmacytes and fibrosis in the local lesions, sometimes accompanied by follicular formation [3] (Fig. 2).

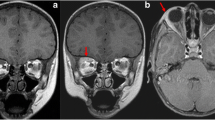
Clinical findings of IgG4-related ophthalmic disease. a Typical IgG4-related ophthalmic disease showing symmetric enlargement of the bilateral lacrimal glands. b Coronal MRI (T1-weighted) depicts marked symmetric enlargement of the bilateral lacrimal glands. c Coronal MRI (T2-weighted) depicts moderate enlargement of the bilateral lacrimal glands and some extraocular muscles and marked enlargement of the infraorbital nerve (trigeminal nerve) (arrow)

Histopathologic findings of IgG4-related ophthalmic disease. a Lymphoplasmacytic infiltration accompanied by follicular formation and marked fibrosis. H&E staining. Bar = 500 µm. b Magnified image of a follicle with a germinal center. H&E staining. Bar = 100 µm. c Immunohistochemical staining showing many IgG4-positive cells. Bar = 100 µm
In the field of ophthalmology, patients with symmetric enlargement of the lacrimal and salivary glands, who used to be diagnosed as having Mikulicz disease [4], were found to have abnormally high serum IgG4 levels [5]. Similar cases were subsequently confirmed by many facilities in Japan and other countries [6–12]. Earlier reports described IgG4-related disease involving the ocular adnexa as characterized by symmetrical and persistent swelling of the lacrimal and salivary glands as well as prominent infiltration of IgG4-expressing plasmacytes in those glands [5–8]. However, it is now known that IgG4-related ophthalmic lesions are not limited to the lacrimal glands but may also manifest diffusely in the orbit and in diverse ocular tissues such as the extraocular muscles, orbital nerve (trigeminal nerve branch), and eyelid [13]. In particular, enlargement of the infraorbital or supraorbital nerve, or both, is frequently observed in this disease [13–15].
Mikulicz disease was first reported in the 1880s [4]. Approximately 50 years later, when Sjögren syndrome was established as a disease entity, Mikulicz disease was considered to be histopathologically equivalent to Sjögren syndrome [16]. However, the differences in the histopathology and lacrimal secretion of the two diseases had been pointed out [17] even before the concept of IgG4-related disease was established.
Names for IgG4-related ocular lesions
Various names have been employed to describe IgG4-related disease. In the field of ophthalmology, terms used include “ocular adnexal IgG4-related disease” [7], “IgG4+ chronic sclerosing dacryoadenitis” [18], “IgG4-related orbital inflammation” [19], “ocular adnexal IgG4-related disease” [20], “IgG4-related dacryoadenitis” [21], and “IgG4-related Mikulicz’s disease” [22].
However, since 2012, following publication of the comprehensive diagnostic criteria for IgG4-related disease [1] in Japan, “IgG4-related ophthalmic disease” has been advocated as the standardized name for the ocular lesions of this disease.
Diagnostic criteria for IgG4-related ophthalmic disease, 2014
As described above, the comprehensive diagnostic criteria of IgG4-related disease for all systemic conditions were reported in 2012 [1]. However, other than for the kidney [23] and pancreas [24], the diagnostic criteria for individual organs are not well established. Against this background, the Study Group for IgG4-related Disease in the Intractable Disease Project supported by a grant from the Ministry of Health, Labor, and Welfare of Japan has played a central role in developing new diagnostic criteria for IgG4-related ophthalmic disease (Table 2). Essentially, the diagnostic criteria are based on the framework of the “Comprehensive Diagnostic Criteria for IgG4-related Disease 2011” (Table 1) [1], which was originally developed mainly for internal medicine, with emphasis given to the specific features of the ophthalmic lesions. In practice, diagnosis is classified as definitive, probable, or possible.
The present criteria and the “Comprehensive Diagnostic Criteria for IgG4-related Disease 2011” [1] differ in several aspects. First, the ocular manifestations including enlargement of the lacrimal gland, trigeminal nerve (supraorbital and infraorbital nerves), extraocular muscle, and various ophthalmic tissues are described in more detail to reflect the involvement of various ocular tissues [13]. Second, in the ocular adnexa, fibrosis is not necessarily marked histopathologically. Third, a germinal center is frequently observed. Fourth, the criteria for an IgG4-positive plasma cell infiltrate is a ratio of IgG4-positive cells to IgG-positive cells of 40 % or above, or 50 or more cells per high-power field (×400). Fifth, the diseases requiring differentiation include Sjögren syndrome and diseases involving tumors or hypertrophic lesions in the ocular adnexa, such as lymphoma and sarcoidosis (Table 3). In addition, attention is drawn to the differentiation from mucosa-associated lymphoid tissue (MALT) lymphoma, which is the most common lymphoproliferative disease in the orbit. The relationship between IgG4-related disease and MALT lymphoma in the ocular adnexa has been reported [25–27] and should be discussed in the future.
Prevalence of IgG4-related ophthalmic disease
Not only is the entity of IgG4-related ophthalmic disease not well recognized and not widely known, the prevalence of the disease also remains unclear. According to a recent multicenter survey in Japan, among 1014 cases of orbital lymphoproliferative disease, MALT lymphoma had the highest prevalence (39.8 %), followed by IgG4-related ophthalmic disease (21.6 %) [28]. Together, these two diseases occupy over 60 % of all cases of lymphoproliferative disease in the orbit.
Among the ophthalmologic cases treated repeatedly with corticosteroids under a clinical diagnosis of “orbital pseudotumor,” “inflammatory pseudotumor of the orbit,” or “idiopathic orbital inflammation,” the possibility exists that a considerable number are in fact IgG4-related ophthalmic disease. Furthermore, among other cases treated under a diagnosis of “orbital myositis,” “orbital apex syndrome,” “idiopathic optic neuropathy,” or “posterior scleritis,” some cases may be difficult to differentiate from IgG4-related ophthalmic disease.
Relationship with lesions in other organs
Although IgG4-related disease lesions may develop in various organs of the body as mentioned above, they do not necessarily occur synchronously in multiple organs. On the contrary, it is not uncommon to find no abnormalities in other organs at the time the ocular lesion is detected and diagnosed. On the other hand, a report has indicated that markedly elevated serum IgG4 (900 mg/dl or higher) is associated with a higher possibility of concurrent orbital and extraorbital lesions, such as in the salivary glands or lymph nodes [29].
Perspective
This review has introduced the diagnostic criteria for a new disease entity, IgG4-related ophthalmic disease. Further evaluation is expected to validate these criteria. In addition, attention has to be paid to the fact that the ocular symptoms of this disease are diverse and, in some cases, may seriously affect visual function.
This disease group is characterized by both temporal and spatial multiplicity of lesion onset and by relatively good response to systemic corticosteroids. However, the lesions may relapse repeatedly following tapering, and progression to a chronic state is not uncommon. On the basis of the diagnostic criteria described in this article, further studies are necessary to develop a classification system of severity specifically for ophthalmic lesions, considering also the clinical findings and the effect on visual function, as well as to establish treatment guidelines centering on corticosteroids. At the same time, through promotion of research on the pathogenesis of IgG4-related ophthalmic disease, elucidation of its etiology may be anticipated.
References
Umehara H, Okazaki K, Masaki Y, Masaki Y, Kawano M, Yamamoto M, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4RD), 2011. Mod Rheumatol. 2012;22:21–30.
Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–8.
Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64:3061–7.
Mikulicz J. Über eine eigenartige symmetrische Erkrankung der Tränen- und Mundspeicheldrüsen. Beitr Z Chir Festschr Theodor Billroth. 1892;2:610–30.
Yamamoto M, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, Takahashi H, et al. Elevated IgG4 concentrations in serum of patients with Mikulicz’s disease. Scand J Rheum. 2004;33:432–3.
Yamada K, Kawano M, Inoue R, Hamano R, Kakuchi Y, Fujii H, et al. Clonal relationship between infiltrating immunoglobulin G4 (IgG4)-positive plasma cells in lacrimal glands and circulating IgG4-positive lymphocytes in Mikulicz’s disease. Clin Exp Immunol. 2008;152:432–9.
Sato Y, Ohshima K, Ichimura K, Sato M, Yamadori I, Tanaka T, et al. Ocular adnexal IgG4-related disease has uniform clinicopathology. Pathol Int. 2008;58:465–70.
Masaki Y, Sugai S, Umehara H. IgG4-related diseases including Mikulicz’s disease and sclerosing pancreatitis: diagnostic insights. J Rheumatol. 2010;37:1380–5.
Kubota T, Moritani S, Katayama M, Terasaki H. Ocular adnexal IgG4-related lymphoplasmacytic infiltrative disorder. Arch Ophthalmol. 2010;128:577–84.
Plaza JA, Garrity JA, Dogan A, Ananthamurthy A, Witzig TE, Salomão DR. Orbital inflammation with IgG4-positive plasma cells: manifestation of IgG4 systemic disease. Arch Ophthalmol. 2011;129:421–8.
Wallace ZS, Khosroshahi A, Jakobiec FA, Deshpande V, Hatton MP, Ritter J, et al. IgG4-related systemic disease as a cause of “idiopathic” orbital inflammation, including orbital myositis, and trigeminal nerve involvement. Surv Ophthalmol. 2012;57:26–33.
Deschamps R, Deschamps L, Depaz R, Coffin-Pichonnet S, Belange G, Jacomet PV, et al. High prevalence of IgG4-related lymphoplasmacytic infiltrative disorder in 25 patients with orbital inflammation: a retrospective case series. Br J Ophthalmol. 2013;97:999–1004.
Sogabe Y, Ohshima K, Azumi A, Takahira M, Kase S, Tsuji H, et al. Location and frequency of lesions in patients with IgG4-related ophthalmic diseases. Graefes Arch Clin Exp Ophthalmol. 2014;52:531–8.
Sogabe Y, Miyatani K, Goto R, Ishii G, Ohshima K, Sato Y. Pathological findings of infraorbital nerve enlargement in IgG4-related ophthalmic disease. Jpn J Ophthalmol. 2012;56:511–4.
Hardy TG, McNab AA, Rose GE. Enlargement of the infraorbital nerve: an important sign associated with orbital reactive lymphoid hyperplasia or immunoglobulin G4-related disease. Ophthalmology. 2014;121:1297–303.
Morgan WS, Castleman B. A clinicopathologic study of “Mikulicz’s disease”. Am J Pathol. 1953;29:471–503.
Tsubota K, Fujita H, Tsuzaka K, Takeuchi T. Mikulicz’s disease and Sjögren’s syndrome. Invest Ophthalmol Vis Sci. 2000;41:1666–73.
Cheuk W, Yuen HK, Chan AC, Shih LY, Kuo TT, Ma MW, et al. Ocular adnexal lymphoma associated with IgG4+ chronic sclerosing dacryoadenitis: a previously undescribed complication of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:1159–67.
Takahira M, Ozawa Y, Kawano M, Zen Y, Hamaoka S, Yamada K, et al. Clinical aspects of IgG4-related orbital inflammation in a case series of ocular adnexal lymphoproliferative disorders. Int J Rheumatol. 2012;2012:635473. doi:10.1155/2012/635473.
Go H, Kim JE, Kim YA, Chung HK, Khwarg SI, Kim CW, et al. Ocular adnexal IgG4-related disease: comparative analysis with mucosa-associated lymphoid tissue lymphoma and other chronic inflammatory conditions. Histopathology. 2012;60:296–312.
Maehara T, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Tanaka A. Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Ann Rheum Dis. 2012;71:2011–9.
Matsui S, Taki H, Shinoda K, Suzuki K, Hayashi R, Tobe K, et al. Respiratory involvement in IgG4-related Mikulicz’s disease. Mod Rheumatol. 2012;22:31–9.
Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, Hisano S, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15:615–26.
The Japan Pancreas Society, the Ministry of Health and Welfare Investigation Research Team for Intractable Pancreatic Disease. Clinical diagnostic criteria for autoimmune pancreatitis 2011 (proposal). Suizo. 2012;27:17–25 (in Japanese).
Sato Y, Takata K, Ichimura K, Tanaka T, Morito T, Tamura M, et al. IgG4-producing marginal zone B-cell lymphoma. Int J Hematol. 2008;88:428–33.
Matsuo T, Ichimura K, Yoshino T. Local recurrence as immunoglobulin G4 (IgG4)-related disease 10 years after radiotherapy to ocular adnexal extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue. J Clin Exp Hematop. 2011;51:125–33.
Kase S, Noda M, Ishijima K, Yamamoto T, Hatanaka K, Ishida S. IgG4-related inflammation of the orbit simulating malignant lymphoma. Anticancer Res. 2013;33:2779–83.
Japanese study group of. IgG4-related ophthalmic disease. A prevalence study of IgG4-related ophthalmic disease in Japan. Jpn J Ophthalmol. 2013;57:573–9.
Kubota T, Katayama M, Moritani S, Yoshino T. Serologic factors in early relapse of IgG4-related orbital inflammation after steroid treatment. Am J Ophthalmol. 2013;155:373–9.
Acknowledgments
This work was supported by the Research Program for Intractable Disease (IgG4-related disease), Health and Labor Sciences Research Group of the Ministry of Health, Labor, and Welfare of Japan.
Conflicts of interest
H. Goto, None; M. Takahira, None; A. Azumi, None.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
The members of the Japanese Study Group for IgG4-Related Ophthalmic Disease are mentioned in the “Appendix.”
Appendix
Appendix
The members of the Japanese Study Group for IgG4-related Ophthalmic Disease are as follows:
Atsushi Azumi, Department of Ophthalmology, Kobe Kaisei Hospital.
Hiroshi Goto, Department of Ophthalmology, Tokyo Medical University.
Kazuko Kitagawa, Department of Ophthalmology, Kanazawa Medical University.
Toshinobu Kubota, Department of Ophthalmology, National Hospital Organization, Nagoya Medical Center.
Yoko Ogawa, Department of Ophthalmology, Keio University School of Medicine.
Koh-ichi Ohshima, Department of Ophthalmology, National Hospital Organization Okayama Medical Center.
Tokuhide Oyama, Division of Ophthalmology and Visual Sciences, Graduate School of Medical and Dental Sciences, Niigata University.
Keigo Shikishima, Department of Ophthalmology, The Jikei University School of Medicine.
Yuka Sogabe, Department of Ophthalmology, Mitoyo General Hospital.
Shigenobu Suzuki, Department of Ophthalmic Oncology, National Cancer Center Hospital.
Masayuki Takahira, Department of Ophthalmology, Kanazawa University Graduate School of Medical Science.
Hideki Tsuji, Department of Ophthalmology, The Cancer Institute Hospital of the Japanese Foundation of Cancer Research.
About this article

Cite this article
Goto, H., Takahira, M., Azumi, A. et al. Diagnostic criteria for IgG4-related ophthalmic disease. Jpn J Ophthalmol 59, 1–7 (2015). https://doi.org/10.1007/s10384-014-0352-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10384-014-0352-2