
Abstract
Purpose
To evaluate the intraocular pressure (IOP)-reducing effects and safety of 0.0015% tafluprost ophthalmic solution (tafluprost) in normal-tension glaucoma (NTG) patients with an IOP of 16 mmHg or less.
Methods
NTG patients with a baseline IOP of 16 mmHg or less were enrolled for a one-eye study in which tafluprost was applied once daily for 12 weeks. The presence of adverse drug reactions and the cumulative incidence of adverse events were also investigated.
Results
Among the 44 enrolled patients, 41/44 (93.2%) eyes completed the study. The baseline IOP was 13.2 ± 1.3 mmHg in the study eyes and 13.0 ± 1.3 mmHg in the fellow eyes, which was not statistically significant (P = 0.9173, Student’s t test). The values obtained for IOP in the study eyes versus fellow eyes were 10.2 ± 1.6 versus 12.1 ± 1.5 mmHg at week 12. The IOP difference between the study eyes and the fellow eyes was statistically significant (P < 0.0001, Student’s t test). The cumulative incidence of adverse events was 58.5% by week 12. Ocular itching was the most frequently observed adverse event (29.3%). All adverse events were clinically tolerable.
Conclusions
Tafluprost induced significant IOP reductions in NTG patients with a baseline IOP of 16 mmHg or less without raising any safety concerns.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Normal-tension glaucoma (NTG) is the most frequently observed type of glaucoma in Japan, where it accounts for 72% of all glaucoma cases [1]. A reduction in intraocular pressure (IOP) is the only evidence-based treatment for glaucoma [2] and is recommended for NTG. Reducing IOP by the use of ophthalmic solutions is preferred for NTG treatment, as it is for other types of glaucoma [3].
Although prostaglandin (PG) analogues are recognized to be the most effective at reducing IOP among a variety of ophthalmic solutions for glaucoma, their efficacy and safety in NTG patients have not been fully investigated [4–6]. Tafluprost is a PGF2α derivative [7, 8] and tafluprost ophthalmic solution (hereafter referred to as “tafluprost”), which was launched in 2008, appears to have an IOP-reducing effect and a safety margin comparable to those of latanoprost in primary open-angle glaucoma (POAG) and ocular hypertension [9–12]. Interestingly, tafluprost is reported to have a significant IOP-reducing effect even in NTG patients with an IOP of 16 mmHg or more [13]. However, no equivalent study has been conducted on NTG patients with an IOP of 16 mmHg or less, even though, according to the Tajimi study, such cases account for 67.5% of NTG in Japan [14].
We therefore designed the present study to evaluate the IOP-reducing effects and safety of tafluprost in NTG patients with a baseline IOP of 16 mmHg or less.
Subjects and methods
Subjects
The subjects were NTG patients more than 20 years old at the time of their hospital visit. The inclusion criteria were as follows: (1) untreated IOP that when measured five times or more within 3 months (baseline IOP) was 16 mmHg or less in both eyes, (2) difference in baseline IOP measurements between the two eyes of <2 mmHg, (3) a visual field defect corresponding to the findings made by examination of the optic nerve head and (4) corrected visual acuity ≥0.7.
The exclusion criteria were as follows: (1) mean deviation values (MD) measured using the Humphrey central 30-2/24-2 SITA standard program of <−15 dB, (2) abnormalities of the anterior segment that could prevent accurate IOP measurement by Goldmann applanation tonometer, (3) a high degree of myopia (spherical equivalent refraction <−10D), (4) cylindrical equivalent refraction <−5D or >+5D, (5) a history of cataract surgery within 1 year and (6) a history of glaucoma operation. Patients who might have used steroid eye drops continuously during this study, who had ocular fundus diseases that might greatly influence the visual field test results, or who were otherwise judged ineligible by the investigator were also excluded from the study. Patients were recruited at the following five institutions: The Jikei University Hospital, Yoshikawa Eye Clinic, Ueno Eye Clinic, Nakano General Hospital and Nihonmatsu Eye Hospital.
Procedures
This was a prospective, multi-center, open-label, one-eye study. The study concepts and other relevant information were fully explained to the potential subjects who met the inclusion criteria (and did not meet the exclusion criteria) according to the principles of the Helsinki Declaration. Among these patients, those who gave informed written consent that was approved by the Ethics Committees of The Jikei University Hospital (IRB no. 20-271 5561) and Nihonmatsu Eye Hospital (IRB no. 20090202-1) were enrolled in this study.
After baseline IOP had been measured five times or more within a period of 3 months, tafluprost (Tapros, Santen, Osaka, Japan) was administered to the eye with the lower MD (the study eye) once daily (1 drop each time) for 12 weeks. The IOP was measured at 2, 4, 8 and 12 weeks after the start of tafluprost instillation. During this study, the IOP of each patient was measured twice at each time point by a single examiner using a Goldmann applanation tonometer. The average of these two IOP measurements was used in the statistical analysis.
During hospital visits throughout the study period, the degree of superficial punctuate keratopathy (SPK) and the degree of conjunctival hyperemia were observed using a slit-lamp microscope. To evaluate the severity of the SPK, the corneal surface was divided into five regions (central, superior, nasal, temporal and inferior), following the report of the National Eye Institute/Industry Workshop [15]. The SPK in each region was scored according to the following system: 0, no punctate staining; 1, sparse punctate staining; 2, dense punctate staining; 3, coalesced, patchy staining; 4, coalesced, planar staining. The score allocated to the most intense staining observed in a given region was used as the score for the whole region.
The conjunctival hyperemia findings were scored on a 4-point scale from 0 through 3, using a standard photograph (Fig. 1). The scores were: 0, no dilatation of vessels was observed; 1, dilatation of mainly small vessels; 2, dilatation of small and large vessels; 3, marked dilatation of small and large vessels. Each adverse event—such as ocular irritation, itching, sensation of a foreign body and dryness—was confirmed and classified by each attending physician using a 5-point scale: nothing, almost nothing, unclear, sometimes or always. Adverse events classed as either sometimes or always were regarded as positive, and counted to determine the cumulative incidence. Changes in the length of eyelashes, exacerbation of iris pigmentation, onset of palpebral pigmentation and deepening of the eyelid sulcus were examined using photographs taken both before and after a 12-week administration.

Photographs used as the standard for scoring conjunctival hyperemia. The findings of conjunctival hyperemia were scored on a 4-point scale from score 0 through score 3. Score 0: no dilatation of vessels is observed; score 1: dilatation of mainly small vessels is observed; score 2: dilatation of small and large vessels is observed; score 3: marked dilatation of small and large vessels is observed
At the start and end of the study, measurements were taken of blood pressure and pulse rates (using an automated sphygmomanometer), as well as of tear meniscus and the Schirmer test (1 min) [16]. To measure the tear meniscus, the tears were stained with fluorescein, and the height at the inferior eyelid margin was examined through the blue filter of a slit-lamp biomicroscope and assessed using a four-grade scale: 0, not observed; 1, linear; 2, narrow; 3, sufficient. The Schirmer test (1 min) was performed as follows: topical anesthetic was applied to the eye and a Schirmer test strip (Color Bar Schirmer Tear Test; Eagle Vision, Memphis, TN) was placed on the inferior eyelid. After 1 min, the extent of wetting was measured in millimeters.
Visual field examination (Humphrey field analyzer SITA standard Program 30-2 or Program 24-2; Carl Zeiss Meditec, Dublin, CA) was performed both before and after the study.
Statistical analysis
All the data obtained at each facility were sent to the data center (Department of Ophthalmology, Jikei University Hospital), and an independent ophthalmologist, one who did not participate directly in the collection of subjects’ data in the study, analyzed them, with personal information being kept confidential.
The difference in IOP between the study eye and the fellow eye in the subjects who could be followed for at least 4 weeks was evaluated using a Student’s t test. The IOP reduction from the baseline IOP was analyzed by repeated-measures analysis of variance (repeated ANOVA). The IOP-reducing effects in the three different baseline IOP groups (11.0–12.5, 12.5–14.0 and 14.0–16.0 mmHg) were investigated by Student’s t test and repeated ANOVA.
Scores given for SPK and hyperemia were analyzed by one-way ANOVA. A paired t test was employed for MD, Schirmer value, meniscus score, blood pressures and pulse rates (after vs. before treatment). The IOP, SPK score, MD, Schirmer value, meniscus score, blood pressure and pulse rate data are presented as means ± standard deviation (SD). Adverse events were analyzed using the cumulative incidence (expressed as a percentage of subjects). Changes in eyelash length, iris pigmentation, palpebral pigmentation and deepening of the eyelid sulcus (after vs. before treatment) were analyzed using the Wilcoxon signed-rank test.
Statistical analysis was performed using JMP version 8.0 software (SAS, Tokyo, Japan), and two-sided P values of <0.05 were considered to be significant.
This study was performed without support from any associations or companies, and none of the authors have any conflicts of interest associated with this study.
Results
This study was conducted from February to October 2009. All 44 patients meeting the inclusion criteria who were enrolled as subjects in the study gave informed consent. After instillation, two patients were lost to follow-up for unknown reasons (completion rate 93.2%). When independent diagnosis was performed by an independent ophthalmologist on the basis of fundus photographs and visual field tests, NTG was confirmed in 41 of the 42 subjects, while optic disc hypoplasia was diagnosed in the remaining one. Consequently, data from 41 eyes in 41 subjects (13 men and 28 women, at a mean age of 59.0 ± 12.5 years), were entered into the data analysis.
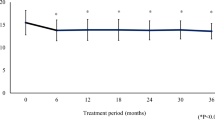
Baseline IOP was 13.2 ± 1.3 mmHg in the study eyes and 13.0 ± 1.3 mmHg in the fellow eyes (not significantly different; P = 0.9173, Student’s t test). The IOP values obtained for the study eyes versus the fellow eyes at weeks 2, 4, 8 and 12 were 10.7 ± 1.8 versus 12.8 ± 1.5 mmHg, 10.3 ± 2.0 versus 12.7 ± 2.2 mmHg, 10.4 ± 1.5 versus 12.4 ± 1.4 mmHg and 10.2 ± 1.6 versus 12.1 ± 1.5 mmHg, respectively (Fig. 2). Each study eye’s value was significantly different from the corresponding fellow eye's value (each at P < 0.0001, Student’s t test). The average IOP differences between the two eyes at weeks 2, 4, 8 and 12 were 0.0 ± 0.6, 2.2 ± 1.0, 2.5 ± 1.3, 2.1 ± 1.1 and 1.9 ± 1.4 mmHg, respectively (Table 1). As compared to the baseline IOP, significant reductions (P < 0.0001, repeated ANOVA) in IOP in the study eyes were observed at weeks 2, 4, 8 and 12. The absolute IOP reductions and percentage IOP reductions from baseline through week 12 are summarized for the study eyes in Table 1. As compared to the baseline level of IOP (13.2 ± 1.3 mmHg), significance was established for the absolute reductions as well as for the percentage reductions from baseline at week 2 (2.5 ± 1.3 mmHg, 19.3 ± 10.2%), week 4 (2.9 ± 1.3 mmHg, 22.2 ± 10.4%), week 8 (2.8 ± 1.2 mmHg, 21.4 ± 9.0%) and week 12 (3.0 ± 1.4 mmHg, 22.7 ± 10.5%) (P < 0.0001, repeated ANOVA; P = 0.0009, 0.0006, 0.0004 and 0.0010, respectively, paired t test).

Time course data for intraocular pressure (IOP) after tafluprost administration. Baseline IOP was 13.2 ± 1.3 mmHg in the study eyes and 13.0 ± 1.3 mmHg in the fellow eyes (not significantly different, P = 0.9173, Student’s t test). The values obtained for IOP in study eyes versus fellow eyes were 10.7 ± 1.8 versus 12.8 ± 1.5 mmHg, 10.3 ± 2.0 versus 12.7 ± 2.2 mmHg, 10.4 ± 1.5 versus 12.4 ± 1.4 mmHg and 10.2 ± 1.6 versus 12.1 ± 1.5 mmHg at weeks 2, 4, 8 and 12, respectively. The average IOP value for the study eyes was significantly decreased (vs. baseline) at weeks 2, 4, 8 and 12 (P < 0.0001, repeated ANOVA). At each time point, the IOP difference between study eyes and fellow eyes was significant (*P < 0.0001, Student’s t test)
When subjects were divided into three groups on the basis of their baseline IOP (11.0–12.5, 12.5–14.0 and 14.0–16.0 mmHg) in the study eyes, the baseline IOP values for the study eyes versus the fellow eyes were 11.9 ± 0.4 versus 11.7 ± 0.5 mmHg (11.0–12.5 mmHg group), 13.0 ± 0.4 versus 13.0 ± 0.6 mmHg (12.5–14.0 mmHg group) and 14.8 ± 0.5 versus 14.6 ± 0.4 mmHg (14.0–16.0 mmHg group). No significant differences were observed between the two eyes in these three groups (mean differences: 0.1 ± 0.4 mmHg, 0.0 ± 0.4 mmHg and 0.1 ± 0.4 mmHg, respectively, P = 0.7054, 0.5221 and 0.5051, respectively, Student’s t test).
The IOP values obtained for the study eyes versus the fellow eyes in the group with an 11.0–12.5 mmHg baseline IOP were 9.5 ± 1.7 versus 11.7 ± 1.2 mmHg, 8.7 ± 1.5 versus 11.0 ± 1.6 mmHg, 9.3 ± 1.3 versus 11.5 ± 1.3 mmHg and 9.2 ± 1.7 versus 11.1 ± 1.6 mmHg at weeks 2, 4, 8 and 12, respectively (Fig. 3), and at each time point the two eyes were significantly different (P = 0.0007, 0.0004, 0.0002 and 0.0071, respectively, Student’s t test). At weeks 2, 4, 8 and 12, the changes in IOP (vs. baseline) in the study eyes were significantly different (P = 0.0005, repeated ANOVA, and P = 0.0002, <0.0001, <0.0001 and 0.0028, paired t test). The values obtained for the average IOP difference between the two eyes at weeks 2, 4, 8 and 12 were 2.2 ± 1.4, 2.3 ± 1.4, 2.2 ± 1.4 and 1.9 ± 1.2 mmHg, respectively (Table 1).

Time course data for IOP in three groups divided according to the baseline IOP in the study eye. No significant difference was observed between the two eyes in baseline IOP in any of these groups (Student’s t test). In study eyes, IOPs were significantly decreased (vs. baseline) at weeks 2, 4, 8 and 12 in each of the three groups (P < 0.001, repeated ANOVA). IOP values were significantly different between the study eyes and the fellow eyes in each group at weeks 2, 4, 8 and 12 (*P < 0.01, Student’s t test)
In the second group, with a 12.5–14.0 mmHg baseline IOP, the IOP values obtained for the study eyes versus the fellow eyes were 10.4 ± 1.4 versus 12.6 ± 1.2 mmHg at week 2, 10.1 ± 1.0 versus 12.8 ± 1.8 mmHg at week 4, 10.3 ± 1.1 versus 12.4 ± 0.8 mmHg at week 8 and 10.2 ± 0.9 versus 12.5 ± 1.2 mmHg at week 12. These values were significantly different between the two eyes (P = 0.0007, <0.0001, <0.0001 and <0.0001, respectively, Student’s t test).
In the third group, with a 14.0–16.0 mmHg baseline IOP, the IOP values obtained for the study eyes versus the fellow eyes were 12.1 ± 1.1 versus 14.2 ± 1.0 mmHg at week 2, 12.3 ± 1.5 versus 14.7 ± 1.3 mmHg at week 4, 11.6 ± 1.4 versus 13.5 ± 0.9 mmHg at week 8 and 11.3 ± 1.3 versus 12.8 ± 1.2 mmHg at week 12. These values were significantly different between the two eyes (P < 0.0001, 0.0004, 0.0007 and 0.0055, respectively, Student’s t test) (Fig. 3). In the groups with 12.5–14.0 and 14.0–16.0 mmHg baseline IOPs, the IOPs in the study eyes were significantly different versus baseline (P = 0.0008 and 0.0046, respectively, repeated ANOVA).
In the 34 (of 41) eyes for which we had all the necessary data, the individual patterns of IOP reductions across the series of visits were divided into four groups (Fig. 4). In group A, the percentage of IOP reduction from baseline was 20% or more from 2 weeks after the start of the administration, and this effect was generally maintained for the rest of the treatment period (10 eyes, 29.4% of the 34 eyes; Fig. 4, group A). In group B, the percentage IOP reduction from baseline was <10% at 12 weeks after the start of the administration, although a reduction in IOP was observed to some extent throughout the treatment period (5 eyes, 14.7% of the 34 eyes; Fig. 4, group B). In group C, the percentage IOP reduction from baseline increased with time between the start of the administration and 12 weeks later, although it fluctuated to some extent during the follow-up (9 cases, 26.5% of the 34 eyes; Fig. 4c). In group D, the percentage IOP reduction from baseline was between 10 and 20% at week 12 and during the follow-up (10 cases, 29.4% of the 34 eyes; Fig. 4d).

Four different patterns of percentage IOP reduction across the 12-week treatment period. Each percentage IOP reduction (vs. baseline) at weeks 2 through 12 was classified as follows: more than 30%, 20–30%, 10–20% and less than 10%. Individual cases displaying similar patterns of percentage IOP reductions are shown grouped together. In group A (10 eyes), the IOP reduction was 20% or more starting from week 2, and this effect was generally maintained throughout the treatment. In group B (5 eyes), the IOP reduction was <10% at week 12, although a reduction in IOP was observed to some extent throughout the treatment. In group C (9 eyes), the IOP reduction tended to increase with time throughout the 12 weeks, although it fluctuated to some extent during the follow-up. In group D (10 eyes), the IOP reduction was over 10 and <20% at week 12 and during the follow-up
Neither the SPK score nor the hyperemia score differed significantly from their baseline values (0.16 ± 0.21 and 0.56 ± 0.74, respectively) throughout the administration period [week 2 (0.22 ± 0.25 and 0.80 ± 0.68), week 4 (0.19 ± 0.31 and 0.63 ± 0.66), week 8 (0.25 ± 0.43 and 0.50 ± 0.61), week 12 (0.22 ± 0.41 and 0.85 ± 0.79)] (P = 0.7863 and P = 0.1363, respectively, one-way ANOVA). No significant difference between the baseline and week 12 values was found for MD (−4.8 ± 4.4 vs. −4.7 ± 4.5 dB, P = 0.1761), the Schirmer value (6.1 ± 5.2 vs. 5.9 ± 5.4 mm, P = 0.4405), the meniscus score (2.0 ± 0.8 vs. 1.9 ± 0.7, P = 0.8263), mean blood pressure (95.2 ± 13.8 vs. 93.2 ± 12.5 mmHg, P = 0.2428) or pulse rate (68.4 ± 10.2 vs. 69.5 ± 11.0 times/min, P = 0.4318).
The cumulative incidence of adverse events during this study was 58.5% by week 12 (24 of 41 subjects). The most frequently observed event was eye itching (29.3%), followed by eye irritation (17.1%), foreign body sensation in the eye (17.1%) and eye dryness (9.8%). Adverse events, including eye itching (29.3%), were observed until week 8 (Fig. 5). Hyperemia (3 of 41 patients), eye mucus (1 of 41 patients) and asthenopia (1 of 41 patients) were observed as transient adverse events. All adverse events were clinically tolerable, and no patients were withdrawn from this study because of adverse events.

Cumulative incidence of adverse events. The cumulative incidence of adverse events in this study was 58.5% (by week 12). The most frequently observed event was eye itching [29.3%, number of patients (pt) = 12], eye irritation (17.1%, pt = 7), foreign-body sensation in eyes (17.1%, pt = 7) and eye dryness (9.8%, pt = 4). Most adverse events (including eye itching) were observed until week 8
Increase in eyelash length between “pre” and 12 weeks after the start of the administration was observed in seven cases (17.1%), which was significant (P = 0.0156, Wilcoxon signed-rank test). Iris pigmentation, palpebral pigmentation and deepening of the eyelid sulcus were observed in 3 cases (7.3%, P = 0.2500), 5 cases (12.2%, P = 0.0625) and 0 cases (0%, P = 1.0000), respectively, none of which was significant (Wilcoxon signed-rank test).
Discussion
The effect of medical treatment on NTG, as well as on other types of glaucoma, is mainly evaluated by the magnitude of the IOP reduction, and so it is important to measure the baseline IOP before treatment by NTG therapy. The objective of this study was to examine the effects of tafluprost on NTG patients with a lower IOP, measurements being taken in each subject five times or more before enrollment to confirm that the baseline IOP was 16 mmHg or less. Subjects were selected only if they had been diagnosed with NTG by their respective physicians, with confirmation by an independent ophthalmologist, on the basis of the optic nerve head findings and visual field examinations. To evaluate the IOP-reducing effects of tafluprost, we conducted one-eye trials as recommended by Japanese clinical guidelines [3]. In such studies, it is important that the IOP of the fellow eye does not alter to an extent sufficient to make judgment of therapeutic efficacy insecure. In this study, using a PG analogue, the IOP difference between the two eyes at baseline was within 2 mmHg, low enough for the results to be valid.
In a previous one-eye trial (of a 12-month administration of travoprost) [17], the travoprost-treated eyes showed a significant IOP reduction versus the control eyes at 3 months after the start of the administration. Although the baseline IOP value was higher in that study than in the present one, in which the baseline IOP was as low as 13.2 ± 1.3 mmHg, the significant drug-induced IOP reductions between the two studies were comparable. Thus, to judge from our data, tafluprost may have a significant IOP-reducing effect in NTG patients with IOP levels of 16 mmHg or less.
A significant IOP reduction (22.4%) was reportedly induced by tafluprost in NTG patients in a phase III randomized controlled trial conducted in Japan [13]. However, a study in which one eye was treated with tafluprost in NTG patients has not previously been reported. Moreover, the subjects of the previous study [13] were not NTG patients with an IOP of 16 mmHg or less. Despite the differences in subjects and in baseline IOP values, the average IOP reduction in our study was around 20%, similar to that in the phase III study [13], which was performed on NTG patients with an IOP of 16 mmHg or more.
In addition, we divided the subjects into three groups according to their mean baseline IOP and compared the IOP reductions among the groups. The IOP levels in the study eyes were significantly different from those in the fellow eyes in each of the three groups at weeks 2, 4, 8 and 12. A favorable percentage of IOP reduction (23.2 ± 14.0% at week 12) was observed even in the group with an 11.0–12.5 mmHg baseline IOP, as well as in the other two groups (12.5–14 mmHg: 21.4 ± 7.8%; 14–16 mmHg: 23.6 ± 9.5%; both at week 12). Thus, tafluprost would seem to have an IOP-reducing effect regardless of the baseline IOP level, even in eyes with an IOP of 16 mmHg or less.
In the study eyes, IOPs were decreased significantly at weeks 2, 4, 8 and 12. The percentage of IOP reduction was 19.3% at week 2 and exceeded 20% at weeks 4, 8 and 12. At week 12, IOP reductions of 20 and 30% were achieved by 61.5 and 20.5% of the eyes, respectively. Although PG analogues induce IOP reductions within a relatively short time after the start of their administration [18, 19], reports of IOP reductions as early as 2 weeks are rare in the clinical setting, and there have been no such reports in NTG patients. Our results indicate that tafluprost may be effective as soon as 2 weeks after the start of administration in NTG patients.
Generally, the IOP-reducing effects of anti-glaucoma ophthalmic solutions are evaluated according to the induced changes in mean IOP, and the individual responses to the drug are not analyzed in detail. Therefore, we decided to examine the time course of the IOP reductions in each subject and to investigate the differences among the subjects in their response to tafluprost. To these ends, the percentage IOP reduction, the time-dependence of the reduction and the duration of the reduction in IOP (all vs. the baseline value) were determined for each eye. We identified four groups. In ten eyes, the IOP reduction was by 20% or more from 2 weeks after the start of administration, and this effect was generally maintained for the remainder of the treatment (Fig. 4a). These subjects would be regarded as good responders to tafluprost. In 5 eyes, the IOP reduction was by <10% at week 12, although reductions in IOP were observed to some extent or other throughout the treatment (Fig. 4b). These subjects could be considered tafluprost non-responders. Non-responders have been reported [20, 21] in studies of other PG analogues, and among those patients some may be non-responders to tafluprost as well. In another nine eyes, the percentage IOP reduction tended to increase with time between the start of the administration and 12 weeks later, although it fluctuated to some extent during the follow-up (Fig. 4c). In these subjects, tafluprost appears to have had a moderate IOP-reducing effect. In ten eyes, the IOP reduction was between 10 and 20% at week 12 and during the follow-up (Fig. 4d). These would appear to be patients who achieve the IOP-reducing effect not at the early stage, but 2–3 months later, as also reported [20] for other PG analogues.
These results support the idea that such late responders may exist for tafluprost treatment as well as for treatment with other PG analogues. Collectively, the above results adequately reflect the clinical features of PG analogues, and suggest that the responses of NTG patients with an IOP of 16 mmHg or less may display similarities among various PG analogues.
In this study, systemic adverse events after instillation of tafluprost were not significant, as also reported for other PG analogues. SPK and conjunctival hyperemia are the most frequent adverse reactions associated with PG analogues [22–25], except eyelash length changes and pigmentation of the iris and palpebra. However, in our study neither the SPK score nor conjunctival hyperemia was found to increase, despite rigorous follow-up. The fact that only a few dry eye patients with a low Schirmer test value or tear meniscus height participated may have affected this outcome. By comparing photographs taken at pre-administration and week 12, eye lashes were found to have significantly increased in length, but the pigmentation of the iris and palpebra and the depth of the eyelid sulcus had not changed significantly. The incidence of eyelash changes in this study was 17.1%, higher than in the previous one-eye study of travoprost (11.11%, indicated as hypertrichosis) [17]. Precise evaluation using photographs may be responsible for this higher incidence. The incidence of latanoprost-induced iris pigmentation was 16.3% at 3 months [17], higher than in the present study (7.3%). Owing to differences in the evaluation and observation methods, these values cannot be compared directly, but values based on slit-lamp photographs were identical between the two studies. These results suggest that physicians should be concerned about possible eyelash changes and pigmentation of the iris and/or palpebra when treating patients with tafluprost, as well as with other PG analogues. Because this result was obtained strictly from evaluation using photographs, the assessment of the severity of the eyelash changes and of the pigmentation of the iris and palpebra was limited to visible signs. Thus, deepening of the eyelid sulcus was not observed in this study.
Adverse events were identified through careful medical interviews and counted to enable assessment of the cumulative incidence of adverse events, which was 58.5%, similar to a rate previously reported (55.1%) [13]. As compared to adverse events reported in a previous study [13], more eye itching, eye irritation and foreign-body sensation in the eyes, but less conjunctival hyperemia and ocular hyperemia were observed in the present study. Even so, all the adverse events were tolerable, and no patient was withdrawn from the study. Most adverse events appeared by week 8, suggesting that physicians should be aware of the possibility of adverse events until at least 8 weeks after the start of tafluprost instillation.
In summary, tafluprost was found to be well tolerated, and significantly reduced IOP in NTG patients with an IOP of 16 mmHg or less. However, the IOP-reducing pattern differed among individual patients. To judge from such patterns, physicians should check the patient’s IOP for at least 3 months before forming an opinion as to the efficacy of tafluprost.
References
Yamamoto T, Iwase A, Araie M, Suzuki Y, Abe H, Shirato S, et al. The Tajimi Study report 2. Ophthalmology. 2005;112:1661–9.
Collaborative Normal-Tension Glaucoma Study Group. The effectiveness of intraocular pressure reduction in the treatment of normal-tension glaucoma. Am J Ophthalmol. 1998;126:498–505.
Japan Glaucoma Society. The Japan glaucoma society guidelines for glaucoma (2nd edition). Nippon Ganka Gakkai Zasshi. 2006;110:777–814.
Tomita G, Araie M, Kitazawa Y, Tsukahara S. A three-year prospective, randomized and open comparison between latanoprost and timolol in Japanese normal-tension glaucoma patients. Eye (London). 2004;18:984–9.
Kondo N, Sawada A, Yamamoto T, Taniguchi T. Correlation between individual differences in intraocular pressure reduction and outflow facility due to latanoprost in normal-tension glaucoma patients. Jpn J Ophthalmol. 2006;50:20–4.
Ang GS, Kersey JP, Shepstone L, Broadway DC. The effect of travoprost on daytime intraocular pressure in normal tension glaucoma: a randomised controlled trial. Br J Ophthalmol. 2008;92:1129–33.
Nakajima T, Matsugi T, Goto W, Kageyama M, Mori N, Matsumura Y, et al. New fluoroprostaglandin F2α derivatives with prostanoid FP-receptor agonistic activity as potent ocular-hypotensive agents. Biol Pharm Bull. 2003;26:1691–5.
Takagi Y, Nakajima T, Shimazaki A, Kageyama M, Matsugi T, Matsumura Y, et al. Pharmacological characteristics of AFP-168 (tafluprost), a new prostanoid FP receptor agonist, as an ocular hypotensive drug. Exp Eye Res. 2004;78:767–76.
Kuwayama Y, Komemushi S. Phase III confirmatory study of 0.0015% DE-085(tafluprost) ophthalmic solution as compared to 0.005% latanoprost ophthalmic solution in patients with open-angle glaucoma or ocular hypertension. Atarashii Ganka. 2008;25:1595–602 (in Japanese).
Uusitalo H, Pillunat LE, Ropo A, On Behalf of the Phase III Study Investigators. Efficacy and safety of tafluprost 0.0015% versus latanoprost 0.005% eye drops in open-angle glaucoma and ocular hypertension: 24-month results of a randomized, double-masked phase III study. Acta Ophthalmol. 2010;88:12–9.
Traverso CE, Ropo A, Papadia M, Uusitalo H. A phase II study on the duration and stability of the intraocular pressure-lowering effect and tolerability of tafluprost compared with latanoprost. J Ocul Pharmacol Ther. 2010;26:97–104.
Egorov E, Ropo A, On Behalf of the Investigators. Adjunctive use of tafluprost with timolol provides additive effects for reduction of intraocular pressure in patients with glaucoma. Eur J Ophthalmol. 2009;19:214–22.
Kuwayama Y, Komemushi S. Intraocular pressure lowering effect of 0.0015% tafluprost as compared to placebo in patients with normal tension glaucoma: randomized, double-blind, multicenter, phase III study. Nippon Ganka Gakkai Zasshi. 2010;114:436–43 (in Japanese).
Iwase A, Suzuki Y, Araie M, Yamamoto T, Abe H, Shirato S, et al. The prevalence of primary open-angle glaucoma in Japanese, The Tajimi Study. Ophthalmology. 2004;111:1641–8.
Yamazaki S, Nanno M, Kimura T, Suzumura H, Yoshikawa K. Effects of switching to SofZia-preserved travoprost in patients who presented with superficial punctate keratopathy while under treatment with latanoprost. Jpn J Ophthalmol. 2010;54:7–14.
Ahmed MB, William GH. One-minute Schirmer test with anesthesia. Cornea. 2003;22:285–7.
Suh MH, Park KH, Kim DM. Effect of travoprost on intraocular pressure during 12 months of treatment for normal-tension glaucoma. Jpn J Ophthalmol. 2009;53:18–23.
Kjellgren D, Douglas G, Mikelberg FS, Drance SM, Alm A. The short-time effect of latanoprost on the intraocular pressure in normal pressure glaucoma. Acta Ophthalmol Scand. 1995;73:233–6.
Mckibbin M, Menage MJ. The effect of once-daily latanoprost on intraocular pressure and pulsatile ocular blood flow in normal tension glaucoma. Eye. 1999;13:31–4.
Ikeda Y, Mori K, Ishibashi T, Naruse S, Nakajima N, Kinoshita S. Latanoprost nonresponders with open-angle glaucoma in the Japanese population. Jpn J Ophthalmol. 2006;50:153–7.
Rossetti L, Gandolfi S, Traverso C, Montanari P, Uva M, Manni G, et al. An evaluation of the rate of nonresponders to latanoprost therapy. J Glaucoma. 2006;15:238–43.
Zimmerman TJ, Stewart WC, Latanoprost Axis Study Group. Intraocular pressure, safety, and quality of life in glaucoma patients switching to latanoprost from monotherapy treatments. J Ocul Pharmacol Ther. 2003;19:405–15.
Kitazawa Y, Latanoprost Study Group. An open-label multicenter study on the efficacy and safety of topical use of latanoprost for 156 weeks. Rinsho Ganka (Jpn J Clin Ophthalmol). 2006;60:2047–54 (in Japanese).
Honrubia F, Garacia-Sanchez J, Polo V, Martinez de la Casa JM, Soto J. Conjunctival hyperaemia with the use of latanoprost versus other prostaglandin analogues in patients with ocular hypertension or glaucoma: a meta-analysis of randomised clinical trials. Br J Ophthalmol. 2009;93:316–21.
Denis P, Baudouin C, Bron A, Nordmann JP, Renard JP, Rouland JF, et al. First-line latanoprost therapy in ocular hypertension or open-angle glaucoma patients: a 3-month efficacy analysis stratified by initial intraocular pressure. BMC Ophthalmol. 2010;10:4.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Nakano, T., Yoshikawa, K., Kimura, T. et al. Efficacy and safety of tafluprost in normal-tension glaucoma with intraocular pressure of 16 mmHg or less. Jpn J Ophthalmol 55, 605–613 (2011). https://doi.org/10.1007/s10384-011-0082-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10384-011-0082-7