
Abstract
Wireworms, the polyphagous larvae of click beetles belonging to the genus Agriotes (Coleoptera: Elateridae), are severe and widespread agricultural pests affecting numerous crops. Biological control agents and methods for this general pest are highly solicited. In a screening for microbial Agriotes pathogens, an intracellular bacterium and a mitosporic fungus were isolated. Phylogenetic analysis based on ribosomal RNA operon sequences of both micro-organisms corroborated their previous morphology-based taxonomic classification. The bacterial pathogen has been assigned to the taxonomic genus Rickettsiella (Gammaproteobacteria) wherein it represents a new pathotype, ‘Rickettsiella agriotidis’, that appears most closely related to subjective synonyms of the nomenclatural type species, Rickettsiella popillae. The fungal pathogen has been shown to belong to the form-species Beauveria bassiana, i.e., an obligate anamorph related to the genus Cordyceps (Ascomycota: Hypocreales). Furthermore, the B. bassiana strain from Agriotes has been shown to be potentially susceptible to identification by gIi-diagnosis, i.e., a diagnostic method making use of the strain-specific presence of self-splicing group-I introns within the ribosomal RNA operons of certain hyphomycetous fungi.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wireworms (Agriotes spp.) are polyphagous subterranean larvae of click beetles (Coleoptera: Elateridae) and, among subterranean arthropods, are the most widespread and most serious agricultural pests worldwide causing severe damage to numerous agricultural crops including maize and potatoes. In Germany and other European countries, the most important wireworm species are Agriotes lineatus (L.), Agriotes obscurus (L.), and Agriotes sputator (L.) (Sufyan et al. 2007; Vidal 2010). A. lineatus and A. obscurus are also reported to cause considerable damage of cereal crops in North America (Vernon et al. 2009). Biological control agents and methods for this general pest are highly solicited.
Rickettsiella bacteria are small, rod-shaped intracellular pathogens of a wide range of arthropods that typically multiply in vacuolar structures within host cells, e.g., of the fat body, and are frequently associated with membrane-bounded protein crystals. The slowly proceeding Rickettsiella infections are cyto- and histologically complex, including polymorphic bacterial development and replication and release from host cells after lysis. Natural transmission is thought to occur perorally by ingestion of released Rickettsiella cells (Tanada and Kaya 1993).
According to the currently valid taxonomy (Fournier and Raoult 2005), Rickettsiella bacteria are named by their original host, i.e., the designation of infra-subspecific pathotypes (with pathotype names written in single quotation marks). Several of these have been placed in synonymy with one of the four currently recognized species, namely the nomenclatural type species Rickettsiella popilliae (Dutky and Gooden), Rickettsiella grylli (Vago and Martoja), Rickettsiella chironomi (Weiser), and Rickettsiella stethorae (Hall and Badgley). The pathotype ‘Rickettsiella melolonthae’, for instance, is considered a subjective synonym of the species R. popilliae. However, further pathotypes await conclusive species assignment.
Due to the resemblance of life cycles, Rickettsiella were originally perceived as “rickettsiae of insects” and had accordingly been assigned to the taxonomic order Rickettsiales (Weiss et al. 1984) that currently belongs to the class Alphaproteobacteria. However, based on 16S rRNA sequencing results from a strain of R. grylli (Roux et al. 1997), the genus Rickettsiella has been reassigned to the order Legionellales of the Gammaproteobacteria, i.e., in comparatively close vicinity to the genera Legionella and Coxiella (Garrity et al. 2005). This reorganization has been confirmed for R. grylli on a genomic basis (Leclerque 2008) and receives support from the determination of 16S rRNA-encoding sequences from further Rickettsiella pathotypes, e.g., from ticks (Kurtti et al. 2002; Leclerque and Kleespies 2012), collembola (Czarnetzki and Tebbe 2004), crustaceans (Cordaux et al. 2007), scarabaeids (Leclerque and Kleespies 2008a; Kleespies et al. 2011), or dipteran insects (Leclerque and Kleespies 2008b), including the Agriotes pathogen (Leclerque et al. 2011) that is studied here in more depth.
Entomopathogenic fungi, e.g., of the genera Metarhizium and Beauveria, have shown promise for the control of soil pests, including wireworms (Butt et al. 2001; Kölliker et al. 2011). The (form-) genus Beauveria comprises an extensive group of filamentous fungal entomopathogens that generally lack a sexual life cycle, but asexually form large numbers of haploid conidiospores that are sympodially arranged on simple conidiophores developing out of basally inflated conidiogenous cells (de Hoog 1972). Today there is broad agreement that Beauveria names a presumably monophyletic group of mostly obligate anamorphs linked to the teleomorphic genus Cordyceps (Ascomycota: Hypocreales: Cordycipitaceae) (Sung et al. 2007). Despite its broad host range and cosmopolitan distribution (Mugnai et al. 1989; Zimmermann 2007), the genus Beauveria has been shown to be comparatively restricted phylogenetically. Species delineation within this taxon has been elucidated by comparison of ribosomal RNA operon internal transcribed spacer (ITS) and elongation factor 1 alpha (EF1a) gene sequences (Rehner and Buckley 2005). Furthermore, Beauveria strains have been shown to contain self-splicing group-I intron sequences in several conserved loci of the 18S and 28S rRNA-encoding genes (Neuvéglise and Brygoo 1994; Coates et al. 2002), a finding that has been exploited in barcoding approaches (Wang et al. 2003) and has been used in the development of a strain-specific identification approaches, termed “gIi-diagnosis”, for Beauveria brongniartii (Neuvéglise et al. 1997; Fatu et al. 2011). In particular, two Beauveria species, B. bassiana and B. brongniartii, have been under intensive study as insect biocontrol agents.
Within the framework of a screening for microbial antagonists of wireworms, dead and diseased larvae and adults from Agriotes spp. from Germany, Switzerland, and Italy were collected and investigated. A detailed overview of the screening program has been published separately (Kleespies et al. 2013). From among the numerous micro-organisms isolated, a Rickettsiella-like intracellular bacterium and a filamentous fungus that had microscopically been identified as a probable member of the genus Beauveria were selected for further study. Here we present a phylogenetic characterization and molecular taxonomic classification based on ribosomal RNA operon sequence comparisons of these two Agriotes pathogens.
Materials and methods
Bacterial DNA was extracted from an infected wireworm that had been collected at Offenbach/Queich (Germany), after surface sterilization with ethanol and removal of a small tissue sample comprising mainly—but not exclusively—fat body tissue, i.e., a presumed major site of replication of Rickettsiella bacteria. The bacterium identified in this wireworm specimen was given the strain designation “JKI E1959/09D”. The selected putative Beauveria strain was isolated from an adult click beetle of the species Agriotes gallicus collected at Ginsheim (Germany) into a single-spore derived pure culture and given the strain designation “JKI E1989”. Fungal genomic DNA was extracted for genetic analysis from this pure culture. In both cases, DNA was extracted using a DNeasy Mini Kit protocol (Qiagen).
PCR amplifications of rRNA operon partial sequences were performed with Phusion High-Fidelity DNA polymerase (New England Biolabs) using the oligonucleotide primers, primer pair specific annealing temperatures (T A), and amplicon specific elongation times (t E) indicated in Table 1 in a reaction comprising an initial denaturation step of 2 min at 94 °C preceding 25 reaction cycles of a 15 s denaturation step at 94 °C, a 30 s annealing step at T A, and an elongation step at 72 °C for t E, followed by a final elongation step of 5 min at 72 °C. All amplifications were performed in three independent reactions from the same genomic DNA template. The quality of PCR products was controlled electrophoretically using ethidium bromide-stained agarose gels, and reactions containing a single appropriately sized product were purified by passage over a Qiaquick PCR purification column (Qiagen) and sequenced on both strands using the fluorescence-labeled dideoxynucleotide technology (SeqLab GmbH, Göttingen, Germany). Raw sequence data were analyzed and combined into consensus sequences using the DNA Strider 1.3 program.
Orthologous sequences available in the GenBank database were identified with the BlastN software tool (Altschul et al. 1997). Sequence alignments were performed by means of the CLUSTAL W function (Thompson et al. 1994) of the MEGA 4 program (Tamura et al. 2007) using an IUB DNA weight matrix. The Tree-Puzzle 5.2 software (Schmidt et al. 2002) was used to estimate dataset-specific parameters. The most appropriate model of DNA sequence evolution was chosen according to the rationale outlined by Posada and Crandall (1998). Organismal phylogenies were reconstructed with the maximum likelihood (ML) method as implemented in the PhyML software tool (Guindon and Gascuel 2003) using the HKY model of nucleotide substitution (Hasegawa et al. 1985) under assumption of a Γ-distribution based model of rate heterogeneity (Yang 1993) allowing for eight rate categories. Tree topology confidence limits were explored in non-parametric bootstrap analyses over 1,000 pseudo-replicates.
Results
In the genetic analysis of the bacterial Agriotes pathogen, strain E1959, an unambiguous consensus sequence was generated from the triplicate raw data for all amplicons. When used as BlastN query, both bacterial 16S and 23S rRNA gene consensus sequences identified best hits from the bacterial genus Rickettsiella.
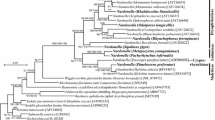
ML phylogenies have been reconstructed for both ribosomal RNA markers (Figs. 1, 2). Importantly, both trees coincide in placing the Agriotes-associated bacterium in maximally bootstrap supported clades comprising, respectively, all 16S and 23S rRNA gene sequences available by 2/2012 from described further Rickettsiella strains.

Bacterial ML phylogeny generated from 16S ribosomal RNA encoding sequences. Terminal branches are labeled by genus, species, pathotype, and strain designations as well as GenBank accession numbers. Numbers on internal branches indicate bootstrap support values. The phylogram has been rooted using Escherichia coli as outgroup. The bar size corresponds to 5 % sequence divergence. To enhance resolution, the upper clade of the phylogram has been extended into a cladogram. ‘R. armadillidii’ partial sequences AM490937-39 show only 68 % sequence coverage with the remaining 16S sequences

Bacterial ML phylogeny generated from 23S ribosomal RNA encoding sequences. Terminal branches are labeled by genus, species, pathotype, and strain designations as well as GenBank accession numbers. Numbers on internal branches indicate bootstrap support values. The phylogram has been rooted using E. coli as outgroup. The bar size corresponds to 5 % sequence divergence. To enhance resolution, the upper clade of the phylogram has been extended into a cladogram
For the fungal Agriotes pathogen, isolate E1989, triplicate raw sequence data gave rise to unambiguous 5.8S rRNA gene and ITS consensus sequences that identified as best hits a plethora of orthologous GenBank entries from the fungal genus Beauveria. In the ML phylogeny that has been reconstructed from a comparison of both ITS and 5.8S rRNA gene sequences from different Beauveria species and further genera of entomopathogenic hyphomycetes (Fig. 3), the new fungal isolate E1989 is firmly placed in a Beauveria clade receiving 94 % bootstrap support and, more exactly, within this clade forms a 98 % bootstrap supported cluster together with strains representing the whole heterogeneity of the species Beauveria bassiana. PCR amplification of the intron insertion regions of both the 18S and 28S rRNA genes from B. bassiana E1989 revealed that there is no intron present in the respective domain of the 18S rRNA gene (data not shown), whereas the size, as estimated from agarose gel elctrophoresis (Fig. 4), of the PCR product obtained from the 28S rRNA-encoding sequence (app. 1.2 kbp) is consistent with the presence of one group-I intron, in contrast to the amplicon sizes expected for the presence of no (app. 0.8 kbp) or two (app. 1.6 kbp) introns.

Fungal ML phylogeny generated from ITS sequences. Terminal branches are labeled by genus, species, and strain designations as well as GenBank accession numbers. To enhance resolution, the upper clade of the phylogram has been extended into a cladogram. Numbers on internal branches indicate bootstrap support values. The phylogram has been rooted using Aspergillus flavus as outgroup. The bar size corresponds to 10 % sequence divergence

Ethidium bromide-stained 1 % agarose gel with fungal PCR products amplified from the 28S rRNA gene intron insertion region using primers I29F and E24R (Table 1). Lanes are labeled as follows: Lane E PCR product from B. bassiana strain E1989 from A. gallicus; lane M 100 bp ladder DNA size standard, with relevant signal sizes being indicated on the left picture margin; lanes 0–2 PCR products from Beauveria standard strains that have previously been shown to carry, respectively, zero, one, or two group-I introns in the 28S rRNA gene
Discussion
For the bacterial Agriotes pathogen, strain E1959, the identification of further available Rickettsiella sequences by the newly determined 23S rRNA gene sequence is in line with its previous 16S rRNA-based identification as representative of a new Rickettsiella pathotype, termed ‘Rickettsiella agriotidis’ (Leclerque et al. 2011), as is the well-supported clade formation in both ribosomal RNA phylogenies (Figs. 1, 2). Somewhat surprisingly, in the case of the 16S rRNA phylogeny, the recently described hypothetical genus ‘Diplorickettsia’ (Mediannikov et al. 2010) is located within the presumed Rickettsiella clade. For both ribosomal RNA markers, ‘R. agriotidis’ E1959 clusters with two R. popilliae—synonymized pathotypes, ‘R. melolonthae’ and ‘Rickettsiella tipulae’, both isolated in Germany, as well as the New Zealandian pathotype ‘Rickettsiella pyronotae’. Relative phylogenetic distances between these four Rickettsiella pathotypes cannot be deduced from ribosomal RNA gene sequence comparisons that are not sufficiently informative as is indicated by the weak bootstrap support values for the respective branches in both the 16S and the 23S rRNA trees. In contrast, ‘R. agriotidis’ E1959 is unambiguously more distantly related to the species R. grylli and appears so with respect to the pathotype ‘Rickettsiella armadillidii’; however, for the latter, no 23S rRNA gene sequence data are available by now. Taking these findings together, phylogenetic reconstruction based on both ribosomal RNA markers corroborates the earlier assignment of the new bacterial Agriotes pathogen to the taxonomic genus Rickettsiella (Gammaproteobacteria: Legionellales). Within this genus, it represents a new pathotype that is accordingly referred to as ‘R. agriotidis’. Moreover, if independently confirmed by more phylogeny informative markers, the comparatively close phylogenetic vicinity to R. popilliae-synonymized pathotypes might motivate placing ‘R. agriotidis’, too, in synonymy to the nomenclatural type species, R. popilliae.
For the fungal Agriotes pathogen, isolate E1989, the identification of BlastN best hits from the genus Beauveria is consistent with its earlier morphologically based characterization as a strain of B. bassiana. This preliminary characterization is fully confirmed by comparison of ITS and 5.8S rRNA gene sequences from different Beauveria species and further genera of entomopathogenic hyphomycetes. Phylogenetic reconstruction together with bootstrap analysis positively characterizes the new fungal isolate as (i) most closely related to the genus Beauveria as compared to further entomopathogenic fungal genera as Isaria, Lecanicillium, or Metarhizium and (ii) most similar to standard strains belonging to the (form-) species B. bassiana, as opposed to further Beauveria species as B. brongniartii, B. pseudobassiana, B. amorpha, B. vermiconia, or B. caledonica. Therefore, the new fungal isolate E1989 from A. gallicus should be considered a strain of B. bassiana.
Moreover, the proven presence of one inserted sequence element in its 28S rRNA gene intron insertion region makes B. bassiana E1989 amenable to the application of a strain-specific gIi-diagnosis strategy using primers complementary to parts of the intron sequence or the intron splice junctions, respectively.
In conclusion, two microbial pathogens of wireworms, Agriotes spp., have been characterized genetically using several ribosomal RNA operon sequences as phylogenetic markers. While a new bacterial pathogen isolated from wireworm has been assigned to the genus Rickettsiella (Gammaproteobacteria) where it forms the first specimen of a new pathotype, ‘R. agriotidis’, a fungal strain isolated from adult A. gallicus turned out to belong to the species B. bassiana. The presence of a group-I intron that might under further investigation be developed into a useful tool of strain-specific molecular identification has been revealed. Bioassays designed to evaluate the biocontrol potential of these wireworm pathogens are currently under way.
References
Altschul SF, Madden TL, Schaffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Butt TM, Jackson C, Magan N (2001) Introduction—fungal biocontrol agents: progress, problems and potential. In: Butt TM, Jackson C, Magan N (eds) Fungi as biocontrol agents: progress, problems and potential. CABI International, Wallingford, pp 1–8
Coates BS, Hellmich RL, Lewis LC (2002) Nuclear small subunit rRNA group I intron variation among Beauveria spp provide tools for strain identification and evidence of horizontal transfer. Curr Genet 41:414–424
Cordaux R, Paces-Fessy M, Raimond M et al (2007) Molecular characterization and evolution of arthropod pathogenic Rickettsiella bacteria. Appl Environm Microbiol 73:5045–5047
Czarnetzki AB, Tebbe CC (2004) Diversity of bacteria associated with Collembola—a cultivation-independent survey based on PCR-amplified 16S rRNA genes. FEMS Microbiol Ecol 49:217–227
de Hoog GS (1972) The genera Beauveria, Isaria, Tritirachium and Acrodontium gen. nov. Stud Mycol 1:1–41
Fournier P-E, Raoult D (2005) Genus II. Rickettsiella Philip, 1956, 267AL. In: Garrity GM, Brenner DJ, Krieg NR et al (eds) Bergey’s manual of systematic bacteriology, vol 2, 2nd edn, part B. Springer, New York, pp 241–247
Garrity GM, Bell JA, Lilburn T (2005) Family II. Coxiellaceae fam. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT (eds) Bergey’s manual of systematic bacteriology. Springer, New York, pp 237–247
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hasegawa M, Kishino H, Yano T-A (1985) Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Kleespies RG, Marshall SDG, Schuster C, Townsend RJ, Jackson TA, Leclerque A (2011) Genetic and electron-microscopic characterization of Rickettsiella bacteria from the manuka beetle, Pyronota setosa (Coleoptera: Scarabaeidae). J Invertebr Pathol 107:206–211
Kleespies RG, Ritter C, Zimmermann G, Burghause F, Feiertag S, Leclerque A (2013) A survey of microbial antagonists of Agriotes wireworms from Germany and Italy. J Pest Sci. doi:10.1007/s10340-012-0447-9
Kölliker U, Biasio L, Jossi W (2011) Potential control of Swiss wireworms with entomopathogenic fungi. IOBC/WPRS Bull 66:517–520
Kurtti TJ, Palmer AT, Oliver JH (2002) Rickettsiella-like bacteria in Ixodes woodi (Acari: Ixodidae). J Med Entomol 39:534–540
Leclerque A (2008) Whole genome-based assessment of the taxonomic position of the arthropod pathogenic bacterium Rickettsiella grylli. FEMS Microbiol Lett 283:117–127
Leclerque A, Kleespies RG (2008a) 16S Ribosomal RNA, GroEL, and MucZ based assessment of the taxonomic position of Rickettsiella melolonthae and its implications for the organization of the genus Rickettsiella. Int J Syst Evol Microbiol 58:749–755
Leclerque A, Kleespies RG (2008b) Genetic and electron-microscopic characterization of Rickettsiella tipulae, an intracellular bacterial pathogen of the crane fly, Tipula paludosa. J Invertebr Pathol 98:329–334
Leclerque A, Kleespies RG (2012) A Rickettsiella bacterium from the hard tick, Ixodes woodi: molecular taxonomy combining multilocus sequence typing (MLST) with significance testing. PLoS ONE 7:e38062
Leclerque A, Kleespies RG, Ritter C, Schuster C, Feiertag S (2011) Genetic and electron-microscopic characterization of ‘Rickettsiella agriotidis’, a new Rickettsiella pathotype associated with a wireworm, Agriotes sp. (Coleoptera: Elateridae). Curr Microbiol 63:158–163
Mediannikov O, Sekeyová Z, Birg M-L, Raoult D (2010) A novel obligate intracellular gamma-proteobacteria associated with Ixodid Ticks, Diplorickettsia massiliensis, Gen. Nov., Sp. Nov. PLoS One 5:e11478
Mugnai L, Bridge PD, Evans HC (1989) A chemotaxonomic evaluation of the genus Beauveria. Mycol Res 92:199–209
Neuvéglise C, Brygoo Y (1994) Identification of group-I introns in the 28S rDNA of the entomopathogenic fungus Beauveria brongniartii. Curr Genet 27:38–45
Neuvéglise C, Brygoo Y, Riba G (1997) 28S rDNA group-I introns: a powerful tool for identifying strains of Beauveria brongniartii. Mol Ecol 6:373–381
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Rehner SA, Buckley E (2005) A Beauveria phylogeny inferred from nuclear ITS and EF1-a sequences: evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia 97:84–98
Roux V, Bergoin M, Lamaze N et al (1997) Reassessment of the taxonomic position of Rickettsiella grylli. Int J Syst Bacteriol 47:1255–1257
Schmidt HA, Strimmer K, Vingron M et al (2002) TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18:502–504
Sufyan M, Neuhoff D, Furlan L (2007) Investigations on click beetles using pheromone traps. Bulletin Oilb/Srop 30:83–87
Sung G-H, Hywel-Jones NL, Sung J-M, Luangsa-ard JJ, Shrestha B, Spatafora JW (2007) Phylogenetic classification of Cordyceps and the clavicipitaceous fungi. Stud Mycol 57:5–59
Tamura K, Dudley J, Nei M et al (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tanada Y, Kaya HK (1993) Insect pathology. Academic Press, San Diego, pp 153–158
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Vernon RS, Van Herk WG, Clodius M et al (2009) Wireworm management I: stand protection versus wireworm mortality with wheat seed treatments. J Econ Entomol 102:2126–2136
Vidal S (2010) Probleme durch neue Schädlinge. Land For 21:17–19
Wang C, Li Z, Typas MA, Butt TM (2003) Nuclear large subunit rDNA group I intron distribution in a population of Beauveria bassiana strains: phylogenetic implications. Mycol Res 107:1189–1200
Weisburg WG, Barns SM, Pelletier DA et al (1991) 16S Ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Weiss E, Dasch GA, Chang K-P (1984) Genus VIII. Rickettsiella Philip 1956. In: Krieg NR, Holt JG (eds) Bergey’s manual of systematic bacteriology. Williams and Wilkins, Baltimore, pp 713–717
White TJ, Bruns TD, Lee SB, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols, a guide to methods and applications. Academic Press, New York, pp 315–322
Yang Z (1993) Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol Biol Evol 10:1396–1401
Zimmermann G (2007) Review on safety of the entomopathogenic fungi Beauveria bassiana and Beauveria brongniartii. Biocontr Sci Technol 17:553–596
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Traugott
Rights and permissions
About this article
Cite this article
Leclerque, A., Mitkovets, P.V., Fatu, AC. et al. Ribosomal RNA phylogeny of bacterial and fungal pathogens of Agriotes wireworms. J Pest Sci 86, 107–113 (2013). https://doi.org/10.1007/s10340-012-0450-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10340-012-0450-1