
Abstract
The Asian citrus psyllid Diaphorina citri (Hemiptera: Psylloidea) is a serious pest of citrus species worldwide because it transmits Candidatus Liberibacter spp. (Alphaproteobacteria: Rhizobiales), the causative agents of the incurable citrus disease, huanglongbing or greening disease. Diaphorina citri possesses a specialized organ called a bacteriome, which harbors vertically transmitted intracellular mutualists, Ca. Carsonella ruddii (Gammaproteobacteria: Oceanospirillales) and Ca. Profftella armatura (Gammaproteobacteria: Betaproteobacteriales). Whereas Carsonella is a typical nutritional symbiont, Profftella is an unprecedented type of toxin-producing defensive symbiont, unusually sharing organelle-like features with nutritional symbionts. Additionally, many D. citri strains are infected with Wolbachia, which manipulate reproduction in various arthropod hosts. In the present study, in an effort to obtain insights into the evolution of symbioses between Diaphorina and bacteria, microbiomes of psyllids closely related to D. citri were investigated. Bacterial populations of Diaphorina cf. continua and Diaphorina lycii were analyzed using Illumina sequencing of 16S rRNA gene amplicons and compared with data obtained from D. citri. The analysis revealed that all three Diaphorina spp. harbor Profftella as well as Carsonella lineages, implying that Profftella is widespread within the genus Diaphorina. Moreover, the analysis identified Ca. Liberibacter europaeus and Diplorickettsia sp. (Gammaproteobacteria: Diplorickettsiales) in D. cf. continua, and a total of four Wolbachia (Alphaproteobacteria: Rickettsiales) lineages in the three psyllid species. These results provide deeper insights into the interactions among insects, bacteria, and plants, which would eventually help to better manage horticulture.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Psyllids or jumping plant lice (Hemiptera: Sternorrhyncha: Psylloidea) are plant sap-sucking insects encompassing about 4000 described species worldwide [1]. They exclusively feed on phloem sap [2], a diet that is deficient in essential amino acids [3] and some vitamins [4, 5]. This nutritional deficiency is compensated for by vertically transmitted intracellular symbionts. Psyllids possess a specialized organ called a bacteriome [6], which typically harbors two distinct bacterial symbionts [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. One is Candidatus Carsonella ruddii (Gammaproteobacteria: Oceanospirillales) [17], which provides the host with essential amino acids that are scarce in the phloem sap [9, 23]. Carsonella is assumed to be present in all psyllid species and is thus categorized as the “primary symbiont” [7,8,9,10,11,12,13,14,15,16,17, 19,20,21,22,23]. Molecular phylogenetic analyses demonstrated congruence between the host and Carsonella trees [12, 16, 17, 20, 21], indicating a single acquisition of an ancestor of Carsonella by a common ancestor of psyllids, followed by strict vertical transmission of symbionts resulting in cospeciation between the host and symbiont lineages. In addition, most psyllid species harbor another bacterial lineage in the bacteriome, which is categorized as a “secondary symbiont” [8, 9, 11, 12, 14, 16, 18, 20]. The secondary symbionts in the psyllid bacteriome are varied depending on psyllid species or genera, suggesting multiple infections and replacements of the symbionts during the evolution of Psylloidea. Whereas the secondary symbionts of various insect lineages have diverse range of associations, from parasitic to mutualistic, with the host [24,25,26,27,28,29,30], those residing in the psyllid bacteriome appear to consistently have obligate mutualistic, organelle-like features like the primary symbionts [9, 11, 12, 14]. Such features are characteristic of nutritional symbionts [24, 31,32,33,34,35,36,37,38,39]. Indeed, whole genome analyses showed that the secondary symbionts of two psyllid species Ctenarytaina eucalypti (Aphalaridae: Spondyliaspidinae) and Heteropsylla cubana (Psyllidae: Ciriacreminae) are nutritional symbionts that complement incomplete amino acid biosynthetic pathways of Carsonella [9]. In contrast, an unprecedented type of secondary symbiont that falls out of this category was found in the Asian citrus psyllid, Diaphorina citri (Liviidae: Euphyllurinae).
Diaphorina citri is an important agricultural pest that transmits Candidatus Liberibacter spp. (Alphaproteobacteria: Rhizobiales), primarily Ca. Liberibacter asiaticus (CLas), the causative agent of a devastating citrus disease known as huanglongbing (HLB) or greening disease [40,41,42]. Diaphorina citri and CLas were originally distributed in tropical and subtropical South to East Asia but were relatively recently introduced into the Arabian Peninsula, Mascarenes, Oceania, and Caribbean, South, Central, and North America [40,41,42]. Because HLB is currently incurable, controlling D. citri as the vector is presently the most crucial part of HLB management [42]. Whereas the association with Liberibacter is transient, D. citri, like other psyllid species, has more intimate and evolutionarily long-lasting relationships with bacteriome-associated bacteria. Along with the primary symbiont Carsonella, D. citri possesses Ca. Profftella armatura (Gammaproteobacteria: Betaproteobacteriales) as a bacteriome-residing secondary symbiont [43, 44]. Profftella is an intracellular resident found in all D. citri individuals across global populations and has a drastically reduced genome of much less than 1 Mb, which is characteristic of bacteriome-associated nutritional symbionts [24, 31, 32, 45]. However, the genome encodes only a few genes required to supplement the host’s diet [43]. Instead, a large part of the genome is devoted to a gene set for synthesizing a secondary metabolite, diaphorin, a polyketide exhibiting significant cytotoxicity to various organisms [43, 46,47,48]. Thus, Profftella is considered to be an unprecedented type of defensive symbiont with organelle-like features. Furthermore, genomic and phylogenetic analyses demonstrated that the Liberibacter lineage has horizontally acquired a gene from the Profftella lineage, showing ecological and evolutionary interactions between the HLB pathogen and the bacteriome symbiont [49]. In addition to these symbionts, many D. citri strains are infected with Wolbachia (Alphaproteobacteria: Rickettsiales) [19, 50,51,52,53,54,55,56,57]. Wolbachia have various effects on arthropod hosts behaving as reproductive manipulators, defensive symbionts, or nutritional symbionts [58,59,60]. Although the role of Wolbachia in D. citri is not known, interactions between Wolbachia and other symbionts, including Carsonella, Profftella, and Liberibacter, are suggested [53, 61,62,63,64].
Diaphorina citri is a member of a species-rich psyllid genus, which currently includes 78 described and many undescribed species restricted to the Old World and mainly distributed in its warm and dry regions, e.g., the Mediterranean Basin, the Sahel region, South and South West Africa, the Middle East, and the arid parts of the Indian subcontinent and Central Asia; these species are associated with many different plant families [65,66,67,68,69,70]. Diaphorina has been formally classified as a member of Liviidae: Euphyllurinae [71], but recent molecular phylogenomic analyses place the genus as sister to Psyllidae or Triozidae, i.e., outside of Liviidae [1]. Except for D. citri, the microbial symbionts of Diaphorina spp. are unknown. Revealing bacterial flora in different Diaphorina spp. would provide deeper insights into the evolution of symbioses between psyllids and bacteria. This would enhance our understanding of D. citri biology, eventually aiding to improve the efficiency of HLB control.
For this purpose, in the present study, we analyzed bacterial populations in two European species, Diaphorina cf. continua and Diaphorina lycii, using the 16S rRNA gene sequencing technique. For comparison, the bacterial flora of D. citri was also analyzed.
Materials and Methods
Insects
Diaphorina cf. continua was collected from Thymelaea tartonraira subsp. thomasii (Thymelaeaceae) in a pine forest 1.8 km west of Moltifao village (42°29′12″N, 9°8′22″E, 300 m.a.s.l.), Haute-Corse department, Corsica island, France, on April 9, 2017. Morphologically, these specimens are similar to D. continua, originally described from Morocco [72]. Diaphorina continua was also recorded from Algeria and Canary Islands (without host plant data) [66] and Sardinia. In Sardinia, which is geographically close to Corsica, D. continua was reported from Thymelaea tartonraira [73, 74], i.e., the same host plant with the material analyzed in this study. However, the adults from Corsica differ from the published descriptions of D. continua [66, 72] in some details, such as the male terminalia. Thus, the species identity of Diaphorina specimens collected on T. tartonraira in Corsica and Sardinia needs to be confirmed by a detailed taxonomic revision.
Diaphorina lycii was collected from Lycium barbarum (Solanaceae) in the floodplain of the Stara river 2.3 km northeast of Byaga village (42°4′45″N, 24°24′11″E, 250 m.a.s.l.), Pazardzhik Region, Bulgaria, on June 18, 2017. Diaphorina lycii is narrowly oligophagous on Lycium spp. and widely distributed in Southern Europe, North Africa, Middle East, Central Asia, and Mongolia [65, 66, 70].
The material of D. citri was used from a laboratory stock free of Ca. Liberibacter spp. The established colony of D. citri, originally collected from Amami Oshima Island, Kagoshima, Japan (28°23′46″N, 129°31′46″E, 5 m.a.s.l.), was maintained on Murraya paniculata (Rutaceae) kept in incubators set at 28 °C with a 16-h light:8-h dark photoperiod.
DNA Extraction
DNA was extracted from whole bodies of adult D. citri (5 males and 5 females), D. cf. continua (3 males and 8 females), and D. lycii (5 males and 5 females) using a DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. The quality of extracted DNA was assessed using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA), and the quantity was assessed using a Qubit 2.0 Fluorometer with a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific).
Construction and Sequencing of Amplicon Libraries
Bacterial populations in D. citri, D. cf. continua, and D. lycii were analyzed using the MiSeq System (Illumina, San Diego, California, USA). The sequencing libraries targeting V3 and V4 regions of the 16S rRNA gene were constructed according to the instructions by Illumina [75] but with some modifications. Briefly, amplicon PCR was performed using the genomic DNA extracted from Diaphorina spp., KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, Massachusetts, USA), and the primer set 16S_341Fmod (5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGYYTAMGGRNGGCWGCAG-3’) and 16S_805R (5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3’). Running parameters were 95 °C for 3 min, followed by 25 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, with a final extension step of 72 °C for 5 min. PCR product sizes were analyzed by agarose gel electrophoresis. Amplicons were purified using Agencourt AMPure XP beads (Beckman Coulter, Brea, California, USA). Dual indices and Illumina sequencing adapters were attached to the amplicons with index PCR using Nextera XT Index Kit v2 (Illumina). Running parameters were 95 °C for 3 min, followed by 8 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s, with a final extension step of 72 °C for 5 min. Amplicons were purified again using Agencourt AMPure XP beads, which were then quantified using a Qubit 2.0 Fluorometer with a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). The libraries were combined with PhiX Control v3 (Illumina), and both ends of 250 bp were sequenced on the MiSeq platform (Illumina) with a MiSeq Reagent Kit v2 (500 cycles; Illumina).
Computational Analysis of Bacterial Populations
Amplicon sequence reads were demultiplexed using MiSeq Reporter (Illumina). The output sequences in FASTQ files per sample were imported into the QIIME2 platform (version 2019.7) [76] and processed using a set of plugins. Primer sequences were removed using the cutadapt plugin [77] with the following options: --p-front-f ^YYTAMGGRNGGCWGCAG --p-front-r ^GACTACHVGGGTATCTAATCC --p-discard-untrimmed. Paired-end sequences were trimmed, denoised, joined, and dereplicated using the dada2 plugin [78] with the following options: --p-max-ee 2 --p-trunc-len-f 230 --p-trunc-len-r 230. During this step, chimeric sequences were detected in samples individually, and sequences found to be chimeric in a sufficient fraction of samples were removed. The q2-feature-classifier plugin [79], a Naive Bayes classifier based on a probabilistic machine learning algorithm, was trained using V3 and V4 regions of 16S rRNA gene sequences in SILVA database ver. 132 (SILVA_132_QIIME_release/taxonomy/16S_only/99/taxonomy_7_levels.txt) that were clustered at 99% sequence similarity. Subsequently, denoised and dereplicated amplicon reads were classified, and taxonomic information was assigned using the trained q2-feature-classifier. Obtained sequence variants (SVs) were manually checked by performing BLASTN searches against the National Center for Biotechnology Information (NCBI) nonredundant (nr) database [80].
Phylogenetic Analysis of Detected Bacteria
SVs after dereplication were aligned with related sequences using SINA (v1.2.11) according to the global SILVA alignment for rRNA genes [81]. Nucleotide sites corresponding to alignment gap(s) were omitted from the data set. Phylogenetic trees were inferred by the maximum likelihood (ML) method using RAxML (version 8.2.12) [82]. The GTR + Γ model was used with no partitioning of the data matrix, with 1000 bootstrap iterations (options -f a -m GTRGAMMA -# 1000).
Results and Discussion
All Three Diaphorina Spp. Have Profftella and Carsonella
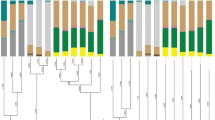
The MiSeq sequencing of the amplicon libraries yielded 90,796 pairs of forward and reverse reads for D. citri, 71,195 paired reads for D. cf. continua, and 32,231 paired reads for D. lycii. Denoising and joining of the paired-end reads along with removal of low-quality or chimeric reads resulted in 69,657 reads for D. citri, 55,673 reads for D. cf. continua, and 30,004 reads for D. lycii (Table S1). Dereplication of these reads resulted in 49 independent SVs, among which only 12 SVs accounted for > 1% of total reads (Table S1). Extremely simple bacterial communities of this type have been reported for sternorrhynchan insects with the bacteriome, including aphids, whiteflies, and other psyllid species [11, 14, 50, 83,84,85,86]. Taxonomic classification by QIIME2 (Fig. 1) followed by independent BLAST searches and phylogenetic analyses (Fig. 2) revealed that all three Diaphorina spp. possess distinct lineages of Profftella. The q2_feature_classifier plugin assigned SV1, SV2, and SV3 to Ca. Profftella armatura. SV1, which was derived from 55.6% of denoised D. citri reads (Table S1), was 100% identical to the corresponding sequence of Profftella previously reported from D. citri populations from Japan (CP003468), China (CP012591), and the USA (EF433792). SV2, which was derived from 40.6% of denoised D. cf. continua reads, was 98.4% identical to SV1. SV3, which was derived from 71.8% of denoised D. lycii reads, was 98.4% and 99.5% identical to SV1 and SV2, respectively. Molecular phylogenetic analysis showed that SV2 and SV3 form a clade with SV1 corresponding to Profftella of D. citri with good bootstrap support (Fig. 2), verifying that both D. cf. continua and D. lycii possess bacterial lineages that are sister to Profftella of D. citri. The finding implies that Profftella lineages are widespread within the genus Diaphorina. Further studies including more psyllid taxa are required to confirm this and to reveal if Profftella is unique to Diaphorina or it also occurs in other psyllid genera.

Composition of bacterial populations in Diaphorina spp. Relative abundances of Illumina reads belonging to assigned bacterial taxa are shown

Phylogenetic position of Profftella lineages within Betaproteobacteriales inferred by the maximum likelihood method. A total of 427 unambiguously aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values over 50% are shown. The scale bar indicates substitutions per site. For symbiotic bacteria, host organisms are shown in brackets. Profftella sequences from this study are shown in bold. DDBJ/EMBL/GenBank accession numbers are provided in parenthesis. Carsonella was used as an outgroup
As expected, the analyses showed that all three Diaphorina spp. possess distinct lineages of Carsonella (Table S1, Figs. 1, S1) that is assumed to be universal in Psylloidea [7,8,9,10,11,12,13,14,15,16,17, 19,20,21,22,23]. The q2_feature_classifier assigned SV4, SV8, and SV11 to Ca. Carsonella ruddii. SV4, which was derived from 30.4% of denoised D. citri reads (Table S1), was 100% identical to the corresponding sequence of Carsonella previously reported from D. citri populations from Japan (CP003467) and China (CP012411) and 99.8% identical to that from the USA (AF211136). SV8, which was derived from 10.8% of denoised D. cf. continua reads, was 99.5% identical to the corresponding sequence of Carsonella previously reported from D. lycii (AF280097) [21] and 98.6% identical to SV4. SV11, which was derived from 4.3% of denoised D. lycii reads, was 99.8% identical to SV8 and the Carsonella sequence previously reported from D. lycii (AF280097) and 98.4% identical to SV4. Phylogenetic analysis showed that SV4, SV8, and SV11 form a strongly supported clade with the Carsonella sequences previously reported from D. citri and D. lycii (Fig. S1), verifying that these SVs correspond to Carsonella lineages. As previously reported [12, 16, 17, 20, 21], the phylogeny of Carsonella showed general congruence with the relationships of their psyllid hosts [1, 87]. Whereas some previous studies that analyzed psyllid microbiomes using “universal primers” detected only a trace amount of Carsonella reads [14, 50, 84], the present study succeeded in detecting a large percentage of Carsonella reads using primers appropriately modified for highly AT-biased symbiont genes [9, 23, 43]. The ratio of Carsonella reads to Profftella reads in D. citri was 0.55, which was consistent with previous reports of quantitative PCR using target-specific primers [51,52,53, 88].
Diaphorina cf. continua has Liberibacter and Diplorickettsia
Taxonomic classification by QIIME2 (Fig. 1) followed by independent BLAST searches and phylogenetic analyses (Fig. 3) identified Ca. Liberibacter europaeus in D. cf. continua. The q2_feature_classifier assigned SV6, which was derived from 23.8% of denoised D. cf. continua reads, to Ca. Liberibacter europaeus (Alphaproteobacteria: Rhizobiales) (Table S1, Fig. 1). SV6 was 100% identical to the corresponding sequence of Ca. Liberibacter europaeus NR-01 (FN678792) and 99.3% identical to Ca. Liberibacter europaeus, isolate Psy6 (JX244258), and isolate BrS (JX244259). Molecular phylogenetic analysis showed that these sequences form a robustly supported clade within Ca. Liberibacter spp. (Fig. 3).

A maximum likelihood phylogram of Liberibacter spp. A total of 402 unambiguously aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values over 50% are shown. For insect-associated bacteria, host insects are shown in brackets. Psyllids are shown in orange. Families and subfamilies of the host psyllids are shown in parenthesis. The sequence from this study is shown in bold. DDBJ/EMBL/GenBank accession numbers for sequences are provided in parenthesis. The scale bar represents nucleotide substitutions per position. Wolbachia was used as an outgroup
The genus Liberibacter currently includes eight species: Ca. L. asiaticus (CLas), Ca. L. americanus (CLam), and Ca. L. africanus (CLaf), which cause HLB in citrus plants (Rutaceae) in Asia, the Americas, and Africa [41, 42]; Ca. L. caribbeanus (CLca) that was identified in citrus in Columbia but the pathogenicity of which is uncertain [89]; Ca. L. solanacearum (CLso), which causes diseases in solanaceous plants in North and Central Americas and New Zealand and in carrot and celery (Apiaceae) in Europe and North Africa [90,91,92,93]; Ca. L. brunswickensis (CLbr), a probable endophyte of solanaceous plants in Australia [94]; L. crescens (Lcr) that is nonpathogenic and the only culturable species in the genus, which was isolated from Babaco papaya (Caricaceae) in Puerto Rico [95]; and Ca. L. europaeus (CLeu) [96,97,98].
CLeu NR-01 (FN678792), which was detected in Cacopsylla spp. (Psyllidae: Psyllinae) and their host rosaceous plants in Italy and Hungary, was described as an endophyte, as it caused no apparent symptoms to the plants [96, 97]. Subsequently, another CLeu lineage (JX244258/JX244259) was found from the broom psyllid Arytainilla spartiophila (Psyllidae: Psyllinae) and its host, the Scotch broom Cytisus scoparius (Fabaceae) with disease symptoms in New Zealand [98]. Arytainilla spartiophila was introduced to New Zealand from the UK for a biological control of the Scotch broom [99]. The CLeu lineage with the sequence (MN176610) identical to that from New Zealand was later confirmed in A. spartiophila and C. scoparius in the UK [100]. The present study adds another example of CLeu from another psyllid species, D. cf. continua, in Corsica island. As field observations suggested that Thymelaea tartonraira (Thymelaeaceae) is the only host plant species for D. cf. continua in Corsica, it would be interesting to assess if this plant species, which is distantly related to C. scoparius (Fabaceae) and rosaceous plants, is also infected with CLeu and if it shows disease symptoms. In the Scotch broom, the presence of CLeu is associated with stunted growth of shoots, shortened internodes, leaf dwarfing, and leaf tip chlorosis [98]. At the moment, data on its possible pathogenicity in T. tartonraira are lacking.
It appears that Ca. Liberibacter lineages have evolved in close associations with Psylloidea, and all known vectors for all Ca. Liberibacter spp. are psyllids. The vectors reported thus far are D. citri for CLas, CLam [41, 42], and CLca [89]; Trioza erytreae (Triozidae) for CLaf [41, 42]; Bactericera cockerelli, B. trigonica, and Trioza apicalis (all Triozidae) for CLso [90, 93]; Acizzia solanicola (Psyllidae: Acizziinae) for CLbr [94]; and Cacopsylla spp. [96, 97], Arytainilla spartiophila [98], and D. cf. continua (this study) for CLeu. Interactions between Ca. Liberibacter spp. and psyllids are assumed to have evolved multiple times independently because of a lack of congruence between the phylogenies of both groups (Fig. 3). This is also the case for associations between Liberibacter and plants. Pelz-Stelinski and Killiny reported that D. citri harboring CLas are more fecund than their uninfected counterparts and overall population fitness of infected psyllids is better [101]. This observed beneficial effect may account for the close associations between Ca. Liberibacter spp. and psyllids. Further studies are required to assess if this hypothesis is applicable to other Ca. Liberibacter -psyllid combinations in general.
The analysis also detected Diplorickettsia sp. (Gammaproteobacteria: Diplorickettsiales) from D. cf. continua. The q2_feature_classifier assigned SV9, which was derived from 9.1% of denoised D. cf. continua reads (Table S1), to Diplorickettsia. SV9 was 98.8% identical to the corresponding sequence of Diplorickettsia sp. MSebKT1 (AB795342), 98.6% identical to the sequence of Diplorickettsia massiliensis 20B (NR_117407), and 97.7% identical to the sequence of Diplorickettsia sp. NS15 (JN606082). Molecular phylogenetic analysis showed that SV9 forms a well-supported clade with these Diplorickettsia spp. (Fig. 4). To our knowledge, this is the first report of Diplorickettsia detected in psyllids.

Phylogenetic position of Diplorickettsia lineages inferred by the maximum likelihood method. A total of 427 unambiguously aligned nucleotide sites were subjected to the analysis. On each branch, bootstrap support values over 50% are shown. The scale bar indicates substitutions per site. For symbiotic bacteria, host organisms are shown in brackets. The sequence from this study is shown in bold, and DDBJ/EMBL/GenBank accession numbers are provided in parenthesis. Carsonella was used as an outgroup
Diplorickettsia massiliensis was first isolated from the European sheep tick Ixodes ricinus (Arachnida: Acari: Ixodidae) collected in Slovakia and proposed to be the type species of a newly described genus Diplorickettsia [102]. The following study detected D. massiliensis from serum samples of human patients with suspected tick-borne disease, suggesting that the bacterium is a human pathogen, like most other bacteria and viruses found in I. ricinus [103]. Diplorickettsia sp. MSebKT1 was found in the leafhopper Macrosteles sexnotatus (Hemiptera: Auchenorrhyncha: Cicadellidae) in Japan, a plant sap-sucking insect that is closely related to psyllids [104]. The clade of Diplorickettsia clustered with Rickettsiella spp. (Gammaproteobacteria: Diplorickettsiales) with a high level of bootstrap support (Fig. 4), corroborating that the genus Diplorickettsia is closely related to the genus Rickettsiella comprising intracellular bacteria that are associated with various arthropods (insects, arachnids, and isopods), including also psyllids [14, 20]. Whereas many Rickettsiella spp. are pathogenic to arthropods, Ca. Rickettsiella viridis [105] found in the aphid Acyrthosiphon pisum (Hemiptera: Sternorrhyncha: Aphidoidea: Aphididae), which is also a close relative of psyllids, alters the aphid body color, potentially affecting the attractiveness of aphids to natural enemies including parasitoids and ladybirds [106]. As little is known about the functions of Diplorickettsia on host arthropods, it would be worth assessing ecological effects of Diplorickettsia to Diaphorina spp., including D. citri.
Four Wolbachia Strains Reside in Diaphorina spp.
Taxonomic classification by QIIME2 (Fig. 1) followed by independent BLAST searches and phylogenetic analyses (Fig. 5) identified four SVs corresponding to distinct lineages of Wolbachia (Alphaproteobacteria: Rickettsiales). Namely, both D. citri and D. lycii were shown to have two strains of Wolbachia, one of which was shared by the two psyllid species, whereas D. cf. continua possessed a single Wolbachia strain that was previously reported from another psyllid genus. SV5 (Wolbachia_i), which was derived from 12.4% of denoised D. citri reads and 16.6% of denoised D. lycii reads (Table S1), was 100% identical to the sequence of Wolbachia previously reported from D. citri in China (GU563890), the planthopper (Hemiptera: Auchenorrhyncha: Delphacidae) Nilaparvata lugens in China (FJ774974) [107], and the aphids (Hemiptera: Sternorrhyncha: Aphidoidea: Aphididae) Phloeomyzus passerinii in China (HQ843849), Cervaphis quercus in China (JN635325), and Cinara cedri in Israel (JN384059) [108]. SV7 (Wolbachia_ii), which was derived from 15.7% of denoised D. cf. continua reads (Table S1), was 100% identical to the sequence of Wolbachia detected from the following insects: the psyllid Bactericera cockerelli (Triozidae) in the USA (KM267305) [109], the whiteflies (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) Bemisia tabaci (MG977008) and Bemisia tuberculata (MG977007) in Brazil, the aphid Cinara cedri in Israel (JN384060) [108], the planthopper Nilaparvata lugens in China (KX280764), the leafhoppers (Hemiptera: Auchenorrhyncha: Cicadellidae) Homalodisca coagulata in the USA (AF501664) [110] and Hishimonoides sellatiformis in Japan (AB073729) [111], the spittlebugs (Hemiptera: Auchenorrhyncha: Aphrophoridae) Philaenus maghresignus in Spain (AB772263) and Aphrophora quadrinotata in the USA (AB772260) [112], the grasshoppers (Orthoptera: Acrididae) Chorthippus parallelus in the Pyrenees (FJ438533) [113] and Stenobothrus lineatus in the UK (EU727131) [114], the mosquito (Diptera: Culicidae) Aedes fluviatilis in Brazil (GQ981315) [115], and the weevil (Coleoptera: Curculionidae) Naupactus cervinus in Brazil (GQ402143) [116]. SV10 (Wolbachia_iii), which was derived from 7.2% of denoised D. lycii reads (Table S1), was 99.8% identical to the sequence of Wolbachia detected in various arthropod hosts including the psyllid Mycopsylla fici (Homotomidae) in Australia (KT273277). SV12 (Wolbachia_iv), which was derived from 1.1% of denoised D. citri reads (Table S1), was 100% identical to the sequence of Wolbachia in D. citri in the USA (EF433793) [117] and Bemisia tabaci in the Philippines (MK157177), Bangladesh (MH370786), Indonesia (KM404233-KM404238), India (KM404186, KM404191, KM404193), Japan (AB981359), China (AY850932, KF454756), and Australia (KF454754).

A maximum likelihood phylogram of Wolbachia. A total of 402 unambiguously aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values over 50% are shown. Host organisms are shown in brackets. The sequence from this study is shown in bold. DDBJ/EMBL/GenBank accession numbers for sequences are provided in parenthesis. Supergroups of Wolbachia are shown in angle brackets. The scale bar represents nucleotide substitutions per position. Liberibacter was used as an outgroup
Wolbachia are rickettsial bacteria widely distributed among various clades of arthropods and nematodes [58,59,60], and the strains are currently classified into supergroups A–Q [118]. Supergroups A and B are monophyletic and are the most common supergroups that infect arthropods, while supergroups C and D infect nematodes. Supergroups E–Q infect a variety of hosts including nematodes, springtails, termites, fleas, aphids, and mites [60]. The molecular phylogenetic analysis in the present study placed SV5, SV7, SV10, and SV12 from Diaphorina spp. in the robustly supported clade of Wolbachia supergroup B (Fig. 5).
Many Wolbachia strains manipulate the reproduction of arthropod hosts through cytoplasmic incompatibility, feminization, male killing, and parthenogenesis, to increase the frequency of infected females in host populations [58,59,60]. With this ability of promoting dissemination, Wolbachia are proposed as promising agents to control insect pests by affecting host traits or microbiomes, including pathogens therein [119, 120]. Because infectious rates of Wolbachia are high in world populations of D. citri [19, 50,51,52,53,54,55,56,57], and interactions between Wolbachia and other symbionts, including Carsonella, Profftella, and Liberibacter, are suggested [53, 61,62,63,64], the application of Wolbachia to control D. citri and/or HLB is anticipated [52, 53, 55, 57, 62]. The present study suggests rampant horizontal transmissions of Wolbachia among various insect lineages including Diaphorina spp., implying that artificial infection and/or removal of Wolbachia are feasible in D. citri. Such techniques would facilitate exploitation of Wolbachia as a tool to control D. citri and/or HLB.
Conclusion
The present study revealed that all three Diaphorina spp. examined harbor Profftella as well as Carsonella lineages, implying that Profftella is widespread within the genus Diaphorina. Moreover, the analysis identified Ca. Liberibacter europaeus and Diplorickettsia sp. in D. cf. continua and a total of four Wolbachia supergroup B lineages in the three psyllid species. These results provide deeper insights into the evolution of interactions among insects, bacteria, and plants, which could eventually help to better manage horticulture.
References
Percy DM, Crampton-Platt A, Sveinsson S, Lemmon AR, Lemmon EM, Ouvrard D, Burckhardt D (2018) Resolving the psyllid tree of life: phylogenomic analyses of the superfamily Psylloidea (Hemiptera). Syst Entomol 43:762–776. https://doi.org/10.1111/syen.12302
Hodkinson ID (1974) The biology of the Psylloidea (Homoptera): a review. Bull Entomol Res 64:325–338
Sandstrom J, Moran N, Sandström J, Moran N (1999) How nutritionally imbalanced is phloem sap for aphids? Entomol Exp Appl 91:203–210
Ziegler H, Pirson A, Zimmermann MH (1975) Nature of transported substances. In: Zimmermann MH, Milburn JA (eds) Transport in plants I. Springer-Verlag, New York, pp 59–100
Nakabachi A, Ishikawa H (1999) Provision of riboflavin to the host aphid, Acyrthosiphon pisum, by endosymbiotic bacteria, Buchnera. J Insect Physiol 45:1–6
Nakabachi A, Koshikawa S, Miura T, Miyagishima S (2010) Genome size of Pachypsylla venusta (Hemiptera: Psyllidae) and the ploidy of its bacteriocyte, the symbiotic host cell that harbors intracellular mutualistic bacteria with the smallest cellular genome. Bull Entomol Res 100:27–33. https://doi.org/10.1017/S0007485309006737
Profft J (1937) Beiträge zur Symbiose der Aphiden und Psylliden. Z Morphol Ökol Tiere 32:289–326
Buchner P (1965) Endosymbiosis of animals with plant microorganisms. Interscience, New York
Sloan DB, Moran NA (2012) Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol Biol Evol 29:3781–3792. https://doi.org/10.1093/molbev/mss180
Arp A, Munyaneza JE, Crosslin JM, Trumble J, Bextine B (2014) A global comparison of Bactericera cockerelli (Hemiptera: Triozidae) microbial communities. Environ Entomol 43:344–352. https://doi.org/10.1603/EN13256
Overholt WA, Diaz R, Rosskopf E et al (2015) Deep characterization of the microbiomes of Calophya spp. (Hemiptera: Calophyidae) gall-inducing psyllids reveals the absence of plant pathogenic bacteria and three dominant endosymbionts. PLoS One 10:1–16. https://doi.org/10.1371/journal.pone.0132248
Hall AAG, Morrow JL, Fromont C, Steinbauer MJ, Taylor GS, Johnson SN, Cook JM, Riegler M (2016) Codivergence of the primary bacterial endosymbiont of psyllids versus host switches and replacement of their secondary bacterial endosymbionts. Environ Microbiol 18:2591–2603. https://doi.org/10.1111/1462-2920.13351
Fromont C, Riegler M, Cook JM (2016) Phylogeographic analyses of bacterial endosymbionts in fig homotomids (Hemiptera: Psylloidea) reveal codiversification of both primary and secondary endosymbionts. FEMS Microbiol Ecol 92:fiw205. https://doi.org/10.1093/femsec/fiw205
Morrow JL, Hall AAG, Riegler M (2017) Symbionts in waiting: the dynamics of incipient endosymbiont complementation and replacement in minimal bacterial communities of psyllids. Microbiome 5:58. https://doi.org/10.1186/s40168-017-0276-4
Fukatsu T, Nikoh N (1998) Two intracellular symbiotic bacteria from the mulberry psyllid Anomoneura mori (Insecta Homoptera). Appl Environ Microbiol 64:3599–3606
Spaulding AW, von Dohlen CD (1998) Phylogenetic characterization and molecular evolution of bacterial endosymbionts in psyllids (Hemiptera: Sternorrhyncha). Mol Biol Evol 15:1506–1513
Thao ML, Moran NA, Abbot P, Brennan EB, Burckhardt DH, Baumann P (2000) Cospeciation of psyllids and their primary prokaryotic endosymbionts. Appl Environ Microbiol 66:2898–2905
Thao ML, Clark MA, Baumann L, Brennan EB, Moran NA, Baumann P (2000) Secondary endosymbionts of psyllids have been acquired multiple times. Curr Microbiol 41:300–304. https://doi.org/10.1007/s002840010138
Subandiyah S, Nikoh N, Tsuyumu S et al (2000) Complex endosymbiotic microbiota of the citrus psyllid Diaphorina citri (Homoptera: Psylloidea). Zool Sci 17:983–989
Spaulding AW, von Dohlen CD (2001) Psyllid endosymbionts exhibit patterns of co-speciation with hosts and destabilizing substitutions in ribosomal RNA. Insect Mol Biol 10:57–67
Thao ML, Clark MA, Burckhardt DH et al (2001) Phylogenetic analysis of vertically transmitted psyllid endosymbionts (Candidatus Carsonella ruddii) based on atpAGD and rpoC: comparisons with 16S-23S rDNA-derived phylogeny. Curr Microbiol 42:419–421
Hansen AK, Jeong G, Paine TD, Stouthamer R (2007) Frequency of secondary symbiont infection in an invasive psyllid relates to parasitism pressure on a geographic scale in California. Appl Environ Microbiol 73:7531–7535
Nakabachi A, Yamashita A, Toh H, Ishikawa H, Dunbar HE, Moran NA, Hattori M (2006) The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science 314:267. https://doi.org/10.1126/science.1134196
Moran NA, McCutcheon JP, Nakabachi A (2008) Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42:165–190. https://doi.org/10.1146/annurev.genet.41.110306.130119
Oliver KM, Degnan PH, Burke GR, Moran NA (2010) Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Annu Rev Entomol 55:247–266. https://doi.org/10.1146/annurev-ento-112408-085305
Johnson KN (2015) The impact of Wolbachia on virus infection in mosquitoes. Viruses 7:5705–5717. https://doi.org/10.3390/v7112903
Ballinger MJ, Perlman SJ (2019) The defensive Spiroplasma. Curr Opin Insect Sci 32:36–41. https://doi.org/10.1016/j.cois.2018.10.004
Wu M, Sun LV, Vamathevan J et al (2004) Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol 2:E69
Degnan PH, Yu Y, Sisneros N, Wing RA, Moran NA (2009) Hamiltonella defensa, genome evolution of protective bacterial endosymbiont from pathogenic ancestors. Proc Natl Acad Sci U S A 106:9063–9068
Degnan PH, Leonardo TE, Cass BN, Hurwitz B, Stern D, Gibbs RA, Richards S, Moran NA (2010) Dynamics of genome evolution in facultative symbionts of aphids. Environ Microbiol 12:2060–2069. https://doi.org/10.1111/j.1462-2920.2009.02085.x
McCutcheon JP, Moran NA (2012) Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10:13–26. https://doi.org/10.1038/nrmicro2670
Moran NA, Bennett GM (2014) The tiniest tiny genomes. Annu Rev Microbiol 68:195–215. https://doi.org/10.1146/annurev-micro-091213-112901
Nakabachi A, Shigenobu S, Sakazume N, Shiraki T, Hayashizaki Y, Carninci P, Ishikawa H, Kudo T, Fukatsu T (2005) Transcriptome analysis of the aphid bacteriocyte, the symbiotic host cell that harbors an endocellular mutualistic bacterium, Buchnera. Proc Natl Acad Sci U S A 102:5477–5482
Nikoh N, McCutcheon JP, Kudo T et al (2010) Bacterial genes in the aphid genome: absence of functional gene transfer from Buchnera to its host. PLoS Genet 6:e1000827
Nikoh N, Nakabachi A (2009) Aphids acquired symbiotic genes via lateral gene transfer. BMC Biol 7:12
Shigenobu S, Richards S, Cree AGG, Morioka M, Fukatsu T, Kudo T, Miyagishima S, Gibbs RA, Stern DL, Nakabachi A (2010) A full-length cDNA resource for the pea aphid, Acyrthosiphon pisum. Insect Mol Biol 19:23–31. https://doi.org/10.1111/j.1365-2583.2009.00946.x
Sloan DB, Nakabachi A, Richards S, Qu J, Murali SC, Gibbs RA, Moran NA (2014) Parallel histories of horizontal gene transfer facilitated extreme reduction of endosymbiont genomes in sap-feeding insects. Mol Biol Evol 31:857–871. https://doi.org/10.1093/molbev/msu004
Nakabachi A, Ishida K, Hongoh Y et al (2014) Aphid gene of bacterial origin encodes protein transported to obligate endosymbiont. Curr Biol 24:R640–R641. https://doi.org/10.1016/j.cub.2014.06.038
Nakabachi A (2015) Horizontal gene transfers in insects. Curr Opin Insect Sci 7:24–29. https://doi.org/10.1016/j.cois.2015.03.006
Halbert SE, Manjunath KL (2004) Asian citrus psyllids (Sternorrhyncha: Psyllidae) and greening disease in citrus: a literature review and assessment of risk in Florida. Fla Entomol 87:330–353
Bové JM (2006) Huanglongbing: a destructive, newly-emerging, century-old disease of citrus. J Plant Pathol 88:7–37
Grafton-Cardwell EE, Stelinski LL, Stansly PA (2013) Biology and management of Asian citrus psyllid, vector of the huanglongbing pathogens. Annu Rev Entomol 58:413–432. https://doi.org/10.1146/annurev-ento-120811-153542
Nakabachi A, Ueoka R, Oshima K et al (2013) Defensive bacteriome symbiont with a drastically reduced genome. Curr Biol 23:1478–1484. https://doi.org/10.1016/j.cub.2013.06.027
Dan H, Ikeda N, Fujikami M, Nakabachi A (2017) Behavior of bacteriome symbionts during transovarial transmission and development of the Asian citrus psyllid. PLoS One 12:e0189779. https://doi.org/10.1371/journal.pone.0189779
McCutcheon JP, Boyd BM, Dale C (2019) The life of an insect endosymbiont from the cradle to the grave. Curr Biol 29:R485–R495. https://doi.org/10.1016/j.cub.2019.03.032
Yamada T, Hamada M, Floreancig P, Nakabachi A (2019) Diaphorin, a polyketide synthesized by an intracellular symbiont of the Asian citrus psyllid, is potentially harmful for biological control agents. PLoS One 14:e0216319. https://doi.org/10.1371/journal.pone.0216319
Nakabachi A, Okamura K (2019) Diaphorin, a polyketide produced by a bacterial symbiont of the Asian citrus psyllid, kills various human cancer cells. PLoS One 14:e0218190. https://doi.org/10.1371/journal.pone.0218190
Nakabachi A, Fujikami M (2019) Concentration and distribution of diaphorin, and expression of diaphorin synthesis genes during Asian citrus psyllid development. J Insect Physiol 118:103931. https://doi.org/10.1016/j.jinsphys.2019.103931
Nakabachi A, Nikoh N, Oshima K, Inoue H, Ohkuma M, Hongoh Y, Miyagishima SY, Hattori M, Fukatsu T (2013) Horizontal gene acquisition of Liberibacter plant pathogens from a bacteriome-confined endosymbiont of their psyllid vector. PLoS One 8:e82612. https://doi.org/10.1371/journal.pone.0082612
Meng L, Li X, Cheng X, Zhang H (2019) 16S rRNA gene sequencing reveals a shift in the microbiota of Diaphorina citri during the psyllid life cycle. Front Microbiol 10:1948. https://doi.org/10.3389/fmicb.2019.01948
Dossi FCA, da Silva EP, Cônsoli FL (2014) Population dynamics and growth rates of endosymbionts during Diaphorina citri (Hemiptera, Liviidae) ontogeny. Microb Ecol 68:881–889. https://doi.org/10.1007/s00248-014-0463-9
Chu CC, Gill TA, Hoffmann M, Pelz-Stelinski KS (2016) Inter-population variability of endosymbiont densities in the Asian citrus psyllid (Diaphorina citri Kuwayama). Microb Ecol 71:999–1007. https://doi.org/10.1007/s00248-016-0733-9
Hosseinzadeh S, Shams-Bakhsh M, Mann M, Fattah-Hosseini S, Bagheri A, Mehrabadi M, Heck M (2019) Distribution and variation of bacterial endosymbiont and “Candidatus Liberibacter asiaticus” titer in the Huanglongbing insect vector, Diaphorina citri Kuwayama. Microb Ecol 78:206–222
Saha S, Hunter WB, Reese J et al (2012) Survey of endosymbionts in the Diaphorina citri metagenome and assembly of a Wolbachia wDi draft genome. PLoS One 7:e50067. https://doi.org/10.1371/journal.pone.0050067
Guidolin AS, Cônsoli FL (2013) Molecular characterization of Wolbachia strains associated with the invasive Asian citrus psyllid Diaphorina citri in Brazil. Microb Ecol 65:475–486. https://doi.org/10.1007/s00248-012-0150-7
Lashkari M, Manzari S, Sahragard A, Malagnini V, Boykin LM, Hosseini R (2014) Global genetic variation in the Asian citrus psyllid, Diaphorina citri (Hemiptera: Liviidae) and the endosymbiont Wolbachia: links between Iran and the USA detected. Pest Manag Sci 70:1033–1040. https://doi.org/10.1002/ps.3643
Chu C, Hoffmann M, Braswell WE, Pelz-Stelinski KS (2019) Genetic variation and potential coinfection of Wolbachia among widespread Asian citrus psyllid (Diaphorina citri Kuwayama) populations. Insect Sci 26:671–682. https://doi.org/10.1111/1744-7917.12566
Stouthamer R, Breeuwer JA, Hurst GD (1999) Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu Rev Microbiol 53:71–102. https://doi.org/10.1146/annurev.micro.53.1.71
Werren JH, Baldo L, Clark ME (2008) Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol 6:741–751. https://doi.org/10.1038/nrmicro1969
Pascar J, Chandler CH (2018) A bioinformatics approach to identifying Wolbachia infections in arthropods. PeerJ 6:e5486. https://doi.org/10.7717/peerj.5486
Jain M, Fleites LA, Gabriel DW (2017) A small Wolbachia protein directly represses phage lytic cycle genes in “Candidatus Liberibacter asiaticus” within psyllids. mSphere 2:e00171–e00117. https://doi.org/10.1128/mSphereDirect.00171-17
Kruse A, Fattah-Hosseini S, Saha S, Johnson R, Warwick E, Sturgeon K, Mueller L, MacCoss M, Shatters RG Jr, Cilia Heck M (2017) Combining ‘omics and microscopy to visualize interactions between the Asian citrus psyllid vector and the Huanglongbing pathogen Candidatus Liberibacter asiaticus in the insect gut. PLoS One 12:e0179531. https://doi.org/10.1371/journal.pone.0179531
Mann M, Fattah-Hosseini S, Ammar E-D et al (2018) Diaphorina citri nymphs are resistant to morphological changes induced by “Candidatus Liberibacter asiaticus ” in midgut epithelial cells. Infect Immun 86:e00889–e00817. https://doi.org/10.1128/IAI.00889-17
Hosseinzadeh S, Ramsey J, Mann M et al (2019) Color morphology of Diaphorina citri influences interactions with its bacterial endosymbionts and ‘Candidatus Liberibacter asiaticus’. PLoS One 14:e0216599. https://doi.org/10.1371/journal.pone.0216599
Loginova M (1978) Novye vidy psillid (Homoptera, Psylloidea) [new species of psyllids (Homoptera, Psylloidea)]. In: Medvedev GS (ed) Novye vidy zhivotnykh [new animal species]. Trudy Zool Inst Akad Nauk SSSR, vol 61, pp 30–123
Burckhardt D (1984) The Mediterranean species of Diaphorina Loew (Homoptera, Psylloidea). Phytophaga 2:1–30
Hollis D (1987) A new citrus-feeding psyllid from the Comoro Islands, with a review of the Diaphorina amoena species group (Homoptera). Syst Entomol 12:47–61. https://doi.org/10.1111/j.1365-3113.1987.tb00547.x
Malenovský I, Burckhardt D (2014) Jumping plant-lice of Socotra Island (Hemiptera: Psylloidea). Acta Entomol Mus Natl Pragae 54(Suppl):23–61
Burckhardt D, Yefremova Z, Yegorenkova E (2015) The jumping plant-louse Diaphorina teucrii sp. nov. (Hemiptera, Liviidae) associated with Teucrium (Lamiaceae) and its parasitoid Tamarixia dorchinae sp. nov. (Hymenoptera, Eulophidae) from the Negev desert, Israel. Zootaxa 3920:463–473 https://doi.org/10.11646/zootaxa.3920.3.5
Ouvrard D (2019) Psyl’list – the world Psylloidea database. https://www.hemiptera-databases.org/psyllist. Accessed 16 Nov 2019
Burckhardt D, Ouvrard D (2012) A revised classification of the jumping plant-lice (Hemiptera: Psylloidea). Zootaxa 34:1–34
Loginova M (1972) On the fauna of Psylloidea from Morocco (Homoptera). Comment Biol 47:1–37
Rapisarda C (1991) Faunistic and ecological notes on the psyllids of Sardinia. Mem Soc Entomol Ital 69:7–52
Conci C, Rapisarda C, Tamanini L (1993) Annotated catalogue of the Italian Psylloidea. First part. (Insecta Homoptera). Atti Acad Roveretana Agiati, Ser VII 242(2B):33–135
Illumina (2013) 16S metagenomic sequencing library preparation Part#15044223 Rev.B
Bolyen E, Rideout JR, Dillon MR et al (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. https://doi.org/10.1038/s41587-019-0209-9
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17:10–12
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2 : High-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Bokulich NA, Kaehler BD, Rideout J et al (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinformatics 10:421. https://doi.org/10.1186/1471-2105-10-421
Pruesse E, Peplies J, Glöckner FO (2012) SINA : accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829. https://doi.org/10.1093/bioinformatics/bts252
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Nakabachi A, Ishikawa H, Kudo T (2003) Extraordinary proliferation of microorganisms in aposymbiotic pea aphids, Acyrthosiphon pisum. J Invertebr Pathol 82:152–161. https://doi.org/10.1016/S0022-2011(03)00020-X
Nachappa P, Levy J, Pierson E, Tamborindeguy C (2011) Diversity of endosymbionts in the potato psyllid, Bactericera cockerelli (Triozidae), vector of zebra chip disease of potato. Curr Microbiol 62:1510–1520. https://doi.org/10.1007/s00284-011-9885-5
Russell JA, Weldon S, Smith AH, Kim KL, Hu Y, Łukasik P, Doll S, Anastopoulos I, Novin M, Oliver KM (2013) Uncovering symbiont-driven genetic diversity across North American pea aphids. Mol Ecol 22:2045–2059. https://doi.org/10.1111/mec.12211
Jing X, Wong ACN, Chaston JM, Colvin J, McKenzie C, Douglas AE (2014) The bacterial communities in plant phloem-sap-feeding insects. Mol Ecol 23:1433–1444. https://doi.org/10.1111/mec.12637
Cho G, Malenovský I, Lee S (2019) Higher-level molecular phylogeny of jumping plant lice (Hemiptera: Sternorrhyncha: Psylloidea). Syst Entomol 44:638–651. https://doi.org/10.1111/syen.12345
Ramsey JS, Chavez JD, Johnson R, Hosseinzadeh S, Mahoney JE, Mohr JP, Robison F, Zhong X, Hall DG, MacCoss M, Bruce J, Cilia M (2017) Protein interaction networks at the host–microbe interface in Diaphorina citri, the insect vector of the citrus greening pathogen. R Soc Open Sci 4:160545. https://doi.org/10.1098/rsos.160545
Keremane ML, Ramadugu C, Castaneda A, et al (2015) Report of Candidatus Liberibacter caribbeanus, a new citrus- and psyllid-associated Liberibacter from Colombia, South America. In: APS annual meeting 2015:101-O
Lin H, Lou B, Glynn JM et al (2011) The complete genome sequence of “Candidatus Liberibacter solanacearum”, the bacterium associated with potato zebra chip disease. PLoS One 6:e19135. https://doi.org/10.1371/journal.pone.0019135
Nelson W, Fisher T, Munyaneza JE (2011) Haplotypes of “Candidatus Liberibacter solanacearum” suggest long-standing separation. Eur J Plant Pathol 130:5–12. https://doi.org/10.1007/s10658-010-9737-3
Nelson WR, Sengoda VG, Alfaro-Fernandez AO et al (2013) A new haplotype of “Candidatus Liberibacter solanacearum” identified in the Mediterranean region. Eur J Plant Pathol 135:633–639. https://doi.org/10.1007/s10658-012-0121-3
Teresani GR, Bertolini E, Alfaro-fernández A et al (2014) Association of ‘Candidatus Liberibacter solanacearum’ with a vegetative disorder of celery in Spain and development of a real-time PCR method for its detection. Phytopathology 104:804–811
Morris J, Shiller J, Mann R, Smith G, Yen A, Rodoni B (2017) Novel ‘Candidatus Liberibacter’ species identified in the Australian eggplant psyllid, Acizzia solanicola. Microb Biotechnol 10:833–844. https://doi.org/10.1111/1751-7915.12707
Fagen JR, Leonard MT, Coyle JF, McCullough C, Davis-Richardson AG, Davis MJ, Triplett EW (2014) Liberibacter crescens gen. Nov., sp. nov., the first cultured member of the genus Liberibacter. Int J Syst Evol Microbiol 64:2461–2466. https://doi.org/10.1099/ijs.0.063255-0
Raddadi N, Gonella E, Camerota C et al (2011) “Candidatus Liberibacter europaeus” sp. nov. that is associated with and transmitted by the psyllid Cacopsylla pyri apparently behaves as an endophyte rather than a pathogen. Environ Microbiol 13:414–426. https://doi.org/10.1111/j.1462-2920.2010.02347.x
Camerota C, Raddadi N, Pizzinat A et al (2012) Incidence of ‘Candidatus Liberibacter europaeus’ and phytoplasmas in Cacopsylla species (Hemiptera: Psyllidae) and their host/shelter plants. Phytoparasitica 40:213–221
Thompson S, Fletcher JD, Ziebell H et al (2013) First report of “Candidatus Liberibacter europaeus” associated with psyllid infested scotch broom. New Dis Rep 27:6
Syrett P, Fowler SV, Harman HM et al (2007) Establishment of Arytainilla spartiophila Förster (Hemiptera: Psyllidae), a new biological control agent for broom, Cytisus scoparius, in New Zealand. New Zeal Entomol 30:53–62. https://doi.org/10.1080/00779962.2007.9722151
Tannières M, Fowler SV, Manaargadoo-Catin L et al (2020) First report of “Candidatus Liberibacter europaeus” in the United Kingdom. New Dis Reports 41:3. https://doi.org/10.5197/j.2044-0588.2020.041.003
Pelz-Stelinski KS, Killiny N (2016) Better together: association with ‘Candidatus Liberibacter asiaticus’ increases the reproductive fitness of its insect vector, Diaphorina citri (Hemiptera: Liviidae). Ann Entomol Soc Am 109:371–376. https://doi.org/10.1093/aesa/saw007
Mediannikov O, Sekeyova Z, Birg M-L, Raoult D (2010) A novel obligate intracellular gamma-proteobacterium associated with ixodid ticks, Diplorickettsia massiliensis, gen. Nov., Sp. Nov. PLoS One 5:e11478. https://doi.org/10.1371/journal.pone.0011478
Subramanian G, Mediannikov O, Angelakis E, Socolovschi C, Kaplanski G, Martzolff L, Raoult D (2012) Diplorickettsia massiliensis as a human pathogen. Eur J Clin Microbiol Infect Dis 31:365–369. https://doi.org/10.1007/s10096-011-1318-7
Ishii Y, Matsuura Y, Kakizawa S, Nikoh N, Fukatsu T (2013) Diversity of bacterial endosymbionts associated with Macrosteles leafhoppers vectoring phytopathogenic phytoplasmas. Appl Environ Microbiol 79:5013–5022. https://doi.org/10.1128/AEM.01527-13
Tsuchida T, Koga R, Fujiwara A, Fukatsu T (2014) Phenotypic effect of “Candidatus Rickettsiella viridis,” a facultative symbiont of the pea aphid (Acyrthosiphon pisum), and its interaction with a coexisting symbiont. Appl Environ Microbiol 80:525–533. https://doi.org/10.1128/AEM.03049-13
Tsuchida T, Koga R, Horikawa M et al (2010) Symbiotic bacterium modifies aphid body color. Science 330(80):1102–1104. https://doi.org/10.1126/science.1195463
Tang M, Lv L, Jing S, Zhu L, He G (2010) Bacterial symbionts of the brown planthopper, Nilaparvata lugens (Homoptera: Delphacidae). Appl Environ Microbiol 76:1740–1745. https://doi.org/10.1128/AEM.02240-09
Augustinos AA, Santos-Garcia D, Dionyssopoulou E, Moreira M, Papapanagiotou A, Scarvelakis M, Doudoumis V, Ramos S, Aguiar AF, Borges PA, Khadem M, Latorre A, Tsiamis G, Bourtzis K (2011) Detection and characterization of Wolbachia infections in natural populations of aphids: is the hidden diversity fully unraveled? PLoS One 6:e28695. https://doi.org/10.1371/journal.pone.0028695
Cooper WR, Swisher KD, Garczynski SF et al (2015) Wolbachia infection differs among divergent mitochondrial haplotypes of Bactericera cockerelli (Hemiptera: Triozidae). Ann Entomol Soc Am 108:137–145. https://doi.org/10.1093/aesa/sau048
Moran NA, Dale C, Dunbar H et al (2003) Intracellular symbionts of sharpshooters (Insecta: Hemiptera: Cicadellinae) form a distinct clade with a small genome. Environ Microbiol 5:116–126
Mitsuhashi W, Saiki T, Wei W, Kawakita H, Sato M (2002) Two novel strains of Wolbachia coexisting in both species of mulberry leafhoppers. Insect Mol Biol 11:577–584
Koga R, Bennett GM, Cryan JR, Moran NA (2013) Evolutionary replacement of obligate symbionts in an ancient and diverse insect lineage. Environ Microbiol 15:2073–2081. https://doi.org/10.1111/1462-2920.12121
Zabal-Aguirre M, Arroyo F, Bella JL (2010) Distribution of Wolbachia infection in Chorthippus parallelus populations within and beyond a Pyrenean hybrid zone. Heredity (Edinb) 104:174–184. https://doi.org/10.1038/hdy.2009.106
Duron O, Bouchon D, Boutin S et al (2008) The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol 6:27. https://doi.org/10.1186/1741-7007-6-27
Moreira L a, Iturbe-Ormaetxe I, Jeffery J a et al (2009) A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell 139:1268–1278. https://doi.org/10.1016/j.cell.2009.11.042
Rodriguero MS, Confalonieri VA, Guedes JVC, Lanteri AA (2010) Wolbachia infection in the tribe Naupactini (Coleoptera, Curculionidae): association between thelytokous parthenogenesis and infection status. Insect Mol Biol 19:631–640. https://doi.org/10.1111/j.1365-2583.2010.01018.x
Meyer JM, Hoy MA (2008) Molecular survey of endosymbionts in Florida populations of Diaphorina citri (Hemiptera: Psyllidae) and its parasitoids Tamarixia radiata (Hymenoptera: Eulophidae) and Diaphorencyrtus aligarhensis (Hymenoptera: Encyrtidae). Fla Entomol 91:294–304
Lindsey ARI, Bordenstein SR, Newton ILG, Rasgon JL (2016) Wolbachia pipientis should not be split into multiple species: A response to Ramírez-Puebla et al., “Species in Wolbachia? Proposal for the designation of ‘Candidatus Wolbachia bourtzisii’, ‘Candidatus Wolbachia onchocercicola’, ‘Candidatus Wolbachia blaxteri’, ‘Candidatus Wolbachia brugii’, ‘Candidatus Wolbachia taylori’, ‘Candidatus Wolbachia collembolicola’ and ‘Candidatus Wolbachia multihospitum’ for the different species within Wolbachia supergroups”. Syst Appl Microbiol 39:220–222
Flores HA, O’Neill SL (2018) Controlling vector-borne diseases by releasing modified mosquitoes. Nat Rev Microbiol 16:508–518
Brinker P, Fontaine MC, Beukeboom LW, Salles JF (2019) Host, symbionts, and the microbiome: the missing tripartite interaction. Trends Microbiol 27:480–488. https://doi.org/10.1016/j.tim.2019.02.002
Funding
We thank Václav Čermák (Central Institute for Supervising and Testing in Agriculture, Olomouc, Czech Republic) for his help to IM in acquiring the sample of Diaphorina cf. continua. This work was supported by the Japan Society for the Promotion of Science (https://www.jsps.go.jp) KAKENHI grant number 26292174 and research grants from Tatematsu Foundation and Nagase Science and Technology Foundation to AN. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
The nucleotide sequence data are available in the DDBJ/EMBL/GenBank databases under the accession numbers DRR190968–DRR190970 (MiSeq output) and TAAA01000001–TAAA01000013 (dereplicated sequence variants).
Electronic Supplementary Material
Rights and permissions
About this article

Cite this article
Nakabachi, A., Malenovský, I., Gjonov, I. et al. 16S rRNA Sequencing Detected Profftella, Liberibacter, Wolbachia, and Diplorickettsia from Relatives of the Asian Citrus Psyllid. Microb Ecol 80, 410–422 (2020). https://doi.org/10.1007/s00248-020-01491-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-020-01491-z