
Abstract
Metal–organic frameworks (MOFs) are a new class of hybrid inorganic–organic microporous crystalline materials, which possess unique properties such as high surface area, tunable pore size, and good thermal stability. These unique characteristics make MOFs interesting targets for sample pretreatment. In this work, MIL-53 material based on aluminum and containing amine functional groups (NH2-MIL-53(Al)) was synthesized and applied as an efficient sorbent for development of vortex-assisted dispersive micro-solid phase extraction for eight United States Environmental Protection Agency’s priority phenols from aqueous samples prior to analysis by high-performance liquid chromatography with photodiode-array detection. A simple extraction process was designed. The parameters affecting the extraction efficiency, such as amount of sorbent, extraction time, type of desorption solvent and its volume were investigated. The good linearity in the concentration range of 0.0015–10.0000 μg mL−1 with the coefficients of determination of greater than 0.9929, low limits of detection (0.0004–0.0133 μg mL−1) and relative standard deviations of lower than 10% were obtained. The proposed method has been successfully applied to the determination of phenol compounds in different water sample matrices including treated water, waste water, river water, sea water, lake water, drinking water and tap water. In addition, computational simulation was performed to predict the adsorption ability of NH2-MIL-53(Al) towards the studied phenolic compounds. The computational results were in agreement with the experimental studies and it has been proved that NH2-MIL-53(Al) is promising for enrichment of phenolic pollutants.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Industrial processes and human activities generate various kinds of pollutant in the environment that can cause serious damage to ecosystems and human health. Phenol and its derivatives are one of the most toxic water pollutants, which come out from various industries such as the production of paper, detergents, polymers, pharmaceuticals, adhesives, explosives, phenolic resins, and petrochemical products. Owing to their high toxicities even at low concentrations and prevalent presence in environment, the United States Environmental Protection Agency (US EPA) and the European Union (EU) have classified them as priority pollutants in aquatic environment [1, 2]. Consequently, sensitive and reliable analytical methods for monitoring these compound residues are usually required.
To date, diverse separation techniques have been widely used for determination of phenol compounds [3,4,5,6,7,8,9,10,11,12]. High-performance liquid chromatography (HPLC) and gas chromatography (GC) with different detection modes are the most intensively used for phenols determination. The polar features and low volatility of phenol compounds have favored the use of HPLC, because it avoids the need of derivatization processes. However, direct determination of phenols in environmental samples is usually difficult because of their low concentrations (sub μg L−1). Hence, suitable sample preparation methods are still required for isolation of trace amounts of the target substances from complex matrices, clean-up and preconcentration prior to instrumental analysis. For these purposes, several sample preparation procedures, based on both liquid–liquid extraction (LLE) [4, 6, 7, 12] and solid-phase extraction (SPE) [2, 3, 5, 8,9,10,11], have been used incorporating with instrumental techniques for determination of phenol residues. Among these sample pretreatment approaches, SPE has become a widely used technique due to its simplicity and flexibility of the approach.
Dispersive solid-phase extraction (DSPE), which was firstly introduced by Anastassiades et al. [13], has been proposed as a valuable alternative SPE technology. The method exhibits some advantages over conventional SPE such as without sophisticated equipment, less solvent waste, short time requirement, increase surface area between the analytes and sorbent and avoiding channeling or blocking of cartridges or disks, as occurs in traditional SPE. Generally, DSPE is based on four main steps: dispersion of the sorbent in a sample solution containing target analytes, extraction by sorption, separation of the sorbent with retained analytes from the solution, and solvent elution of the analytes [2]. Recently, the analytical chemists have turned to DSPE-based miniaturized technique, micro-DSPE, which is acquiring a lot of attention nowadays from an environment-friendly point of view, due to the impressive decrease in the amounts of sorbents required (by definition: lower than 500 mg of sorbent) [14]. Hence, dispersive micro-solid phase extraction (D-μ-SPE), a miniaturized mode of DSPE based on using milligram amounts of solid sorbent has been introduced [14, 15].
Solid sorbent are important for the SPE-based technique because it contains the active sites to allow the interaction between sorbent and analytes. Generally, the main requirements for properties of solid sorbent include the fast and quantitative adsorption and desorption, a high capacity and high dispersibility in liquid samples. Many different kinds of materials are potential candidates for extraction and preconcentration of various analytes, such as layered double hydroxides (LDHs) [2], molecularly imprinted polymers (MIPs) [3, 5], polymeric sorbent [8], and zeolite NaY [15]. However, most sorbents are intended for specific substances. Therefore, it is conductive to find a kind of high selective and effective adsorbent for the D-μ-SPE of the analytes of interest.
Metal–organic frameworks (MOFs) are a new class of hybrid inorganic–organic microporous crystalline materials constructed from metal ions and organic linkers through coordination bonds and have been popular materials since the beginning of this century [16]. They have received increasing interests due to their unique structures and fascinating chemical properties, including large surface areas, large internal pore volumes, tunable pore sizes, tailorable molecular structures, and catalysis activity. These properties make MOFs promising in diverse applications, such as gas storage [17, 18], separation [19,20,21], catalysis [21, 22], chemical sensing [23], adsorption [24, 25] and sequestration of toxic metals from water [26, 27]. A large number of reports have shown that the MOFs possess great potential in sorption-related fields. The applications of MOFs for D-μ-SPE procedure has also been reviewed [28].
In this work, amine-functionalized frameworks based on well-known MIL family topology, since these are thermally and chemically stable compared to other MOFs [21, 29], is expected to show promising potential for extraction of phenol compounds via hydrogen bonding and π–π interaction. Most of amine-functionalized MOFs studies focused on the CO2 adsorption and separation properties [20, 21, 30]. Previously, NH2-MIL-101(Al) has been reported for adsorption and separation of phenol and p-nitrophenol from aqueous solutions through hydrogen bonding [24]. Luo et al. reported the synthesis of NH2-MIL-53(Al)-incorporated poly(styrene-divinylbenzene-methacrylic acid) monolith column and its application for in-tube solid-phase microextraction of estrogens in human urine [31]. The prepared monolith column showed well properties after more than 100 extraction cycles. However, fabrication of such a monolith column is rather complicated.
Here, we report the synthesis and application of amine-functionalized MOF sorbent for extraction and preconcentration of phenol residues in different water sample matrices for the first time. Development and optimization of a simple and effective vortex-assisted dispersive micro-solid phase extraction (VA-D-µ-SPE) procedure is also proposed. In addition, molecular modeling was used to predict the adsorption ability of the synthesized sorbent towards phenolic compounds. The free binding energies were calculated and the molecular interactions were demonstrated using molecular docking. This work presented the combined study of computational modeling and experimental application in sample preparation technology.
Experimental
Chemicals and Reagents
Eight analytical phenol standards with a purity of ≥ 99% were used. 2,4-Dinitrophenol (2,4-DNP), 4-chloro-3-methylphenol (4-C-3-MP), 2-methyl-4,6-dinitrophenol (2-M-4,6-DNP), and 2,4,6-trichlorophenol (2,4,6-TCP) were supplied by Sigma-Aldrich (Germany). Phenol (Ph) was obtained from Sigma-Aldrich (USA). 4-Nitrophenol (4-NP) and 2,4-dichlorophenol (2,4-DCP) were purchased from Sigma-Aldrich (India). 2-Nitrophenol (2-NP) was supplied by Fluka (China). Stock standard solutions of phenols at concentration of 1000 µg mL−1 were prepared using methanol as the solvent and diluted to 100 μg mL−1 with water. Working solutions were prepared daily from 100 μg mL−1 of stock standard solutions by dilution with water. Deionized water (18.2 MΩ cm) used in all experiments was prepared by a RiOs Type I Simplicity 185 water purification system (Millipore, USA).
2-Aminoterephthalic acid (HO2C-C6H3NH2-CO2H) (99%, Sigma-Aldrich, USA), aluminum chloride hexahydrate (AlCl3∙6H2O) (99%, Sigma-Aldrich, Germany) and N,N-dimethylformamide (≥ 99%, Merck, Germany) were used for synthesis of amine-functionalized MOF sorbent. Acetonitrile (isocratic grade, Merck, China), methanol (gradient grade, Merck, Germany), ethanol (AR grade, Merck, Germany), acetone (AR grade, Qrëc, New Zealand) and glacial acetic acid (AR grade, Carlo Erba, Italy) were used for HPLC separation and VA-D-µ-SPE procedure.
Apparatus and Chromatographic Conditions
The HPLC system (Waters, USA) consisted of an in-line degasser, a 600E quaternary pump, and a Waters 2996 photodiode array (PDA) detector. Empower software was employed to acquire and analyze chromatographic data. The system was equipped with a Rheodyne injector with a 10-μL loop, and a Phenomenex Luna C18 (4.6 mm × 150 mm, 5 μm) (Phenomenex, USA) analytical column. The separation was performed using acetonitrile (solvent A) and 0.1% acetic acid in water (solvent B) as mobile phase. The gradient program was as follows: 0–3 min, 40% solvent A; 4–8 min, 50% solvent A; 9–12 min, 70% solvent A; 13–17 min, 100% solvent A. A re-equilibration period of 2 min with 40% solvent A was carried out between individual runs. The detections were performed at 271 nm for Ph; at 317 nm for 4-NP; at 258 nm for 2,4-DNP; at 276 nm for 2-NP; 281 nm for 4-C-3-MP; at 286 nm for 2,4-DCP; at 266 nm for 2-M-4,6-DNP; and at 288 nm for 2,4,6-TCP.
The X-ray diffraction (XRD) patterns were recorded with an Empyrean X-ray diffractometer (XRD) (PANalytical, The Netherlands) using Cu Kα radiation (λ = 1.5418 Å) over the angular range from 5° to 50°. Fourier transform infrared (FTIR) spectra were recorded on a TENSOR27 infrared scanner (Bruker, Germany) with a resolution of 2 cm−1 and a spectral range of 4000–550 cm−1. The weight loss curves (TG–DTG) were recorded using a simultaneous thermal analyzer (STA) model STA7200 (Hitachi, Japan) from 100 to 600 °C at a heating rate of 10 °C min−1 under nitrogen.
Other instruments were used in the procedure, including a vortex mixer model G560E (50 Hz) (Scientific Industries, USA), a rotavapor model R-200 (Buchi Labortechnik AG, Switzerland), a centrifuge model Z206A (Hermle Labortechnik, Germany) and an oven model UN110 (Memmert, Germany).
Synthesis of Sorbent
The amine-functionalized MOF sorbent was synthesized by means of a solvothermal treatment adapted from the published procedures [21]. Aluminum chloride hexahydrate (0.51 g) and 2-aminoterephthalic acid (0.56 g) were mixed with N,N-dimethylformamide (30 mL) in a Teflon-lined autoclave and then heated at 403 K in an oven under static condition for 72 h. After cooling to the room temperature, the resulting yellow powder was filtered under vacuum and washed with acetone. To remove organic species trapped within the pores, the samples were activated in boiling methanol overnight and stored at 373 K.
VA-D-µ-SPE Procedure
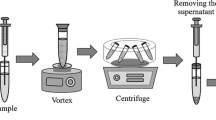
The determination of phenol pollutants was carried out by VA-D-µ-SPE using amine-functionalized MOF as sorbent followed by high-performance liquid chromatography with photodiode-array detection (HPLC-PDA). For this purpose, an aliquot of 10.00 mL aqueous phenol standard or sample solution was added to a 15-mL centrifuge tube containing 30 mg of sorbent. The mixture was then placed in a vortex mixer for 10 s to accelerate the sorption of the target analytes onto the sorbent. Subsequently, the solid sorbent was isolated from the solution by centrifugation at 5000 rpm for 1 min. After that, the supernatant was discarded. Then, 1500 μL of acetonitrile–acetic acid mixture (9.5:0.5, v/v) was added in the centrifuge tube. The analytes were desorbed by vortex mixing for 10 s. The mixture was centrifuged at 5000 rpm for 1 min. The desorption solvent which contained analytes of interest was filtered through a 0.45-μm membrane and evaporated to dryness by rotary evaporator. The residue was reconstituted in 50 μL of acetonitrile before HPLC analysis. The proposed D-µ-SPE-HPLC procedure is schematically depicted in Fig. 1.

Scheme of the proposed VA-D-µ-SPE procedure for determination of phenol pollutants
Sample Collection
Two lake water samples were collected from different areas in Khon Kaen province, Northeastern Thailand (16°24′58.3″N 102°50′07.4″E and 16°26′55.6″N 102°51′03.0″E). Treated water sample (16°28′23.0″N 102°49′07.2″E) and two wastewater samples (16°27′38.3″N 102°48′26.0″E and 16°27′25.6″N 102°48′31.0″E) were collected in Khon Kaen University campus. River water sample was taken from Chi river (Maha Sarakham province, Northeastern Thailand, 16°13′55.0″N 103°15′58.6″E) and seawater sample was taken from Bangsaen beach (Chonburi province, Eastern Thailand, 13°20′32.4″N 100°55′31.0″E). Tap water sample was freshly collected from the laboratory (16°28′33.6″N 102°49′26.6″E) and drinking water samples were bottled water from three different companies sold in Khon Kaen (16°28′39.1″N 102°49′23.7″E). All samples were passed through a Whatman No. 42 filter paper before analysis by the proposed VA-D-µ-SPE-HPLC method.
Molecular Docking
The adsorption ability of MOFs depends directly on the interaction between the analytes and MOFs. In order to investigate such interaction and to understand the adsorption at a molecular level, a molecular docking, a computational technique used to predict the small-molecule conformations adopted within the binding sites of macromolecular targets as well as their interaction, has been performed. The studied amine-functionalized MOF (NH2-MIL-53(Al)) crystal structure was taken from the Cambridge Crystallographic Data Centre (CCDC number: 901254) [32]. All phenolic compound structures were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov) and the structures were re-optimized with quantum calculation B3LYP/6-31G(d) level using Gaussian 09 program [33]. All docking calculations were performed with a grid box size of 60 × 60 × 60 Å3, grid spacing of 0.375 Å, 2.5 × 106 energy evaluations, and 200 runs of the Lamarckian genetic algorithm searches using AutoDock 4.2 program [34]. The binding free energies were calculated and the orientation of the analytes in the binding site of NH2-MIL-53(Al) was investigated.
Results and Discussion
Characterization of the Prepared Sorbent
The synthesized sorbent was characterized by XRD, TGA, and FTIR experiments (Fig. S1, Supplementary Information). The framework structure of as-synthesized sorbent was identified by XRD and the pattern is shown in Fig. S1A. As can be seen, the XRD pattern of the as-synthesized sorbent was in good accordance with the simulated MIL-53, indicating that the pure phase of NH2-MIL-53(Al) was obtained. It has been reported that by starting from the similar synthesis mixture at the same temperature, different MIL structure can be obtained just by changing the synthesis time, 24 h for MIL-101 and 72 h for MIL-53 [22]. This also supports the formation of NH2-MIL-53(Al) in the present work. In order to analyze the molecular structure and identify the functional groups of as-synthesized sorbent, the FTIR spectroscopy was used and the results are shown in Fig. S1B. A series of peaks appeared in the range of 1400–1700 cm−1 were assigned to carboxylic acid that coordinated with A13+ and the carbonyl groups of DMF molecule which adhered to channels of the sorbent. The absorption peaks around 1600 and 1500 cm−1 attributed to asymmetric stretchings of carbonyl, while two peaks at about 1440 and 1400 cm−1 were carbonyl symmetric stretchings. An observed absorption peak at 1670 cm−1 is assigned to stretching vibration of carbonyl group in molecules of free DMF, and the bands at 3500 and 3380 cm−1 are due to the stretching vibration of NH2 group. Therefore, the results from XRD and FTIR analysis clearly confirm the formation of NH2-MIL-53(Al) structure.
The thermal behavior of the synthesized NH2-MIL-53(Al) was studied by TG measurement (Fig. S1C). TGA curve showed a two-step weight loss. First step showed a weight loss of 7% up to 120 °C corresponding to the release of methanol molecules trapped in the pores of the framework structure. The TGA data revealed that the MIL-53 was stable up to 420 °C. At this temperature it showed the starting of a second step weight loss related to the decomposition of organic ligand, aminoterephthalic acid [35].
Optimization of the VA-D-µ-SPE Conditions
The synthesized NH2-MIL-53(Al) was applied as sorbent for development of VA-D-µ-SPE for eight target phenol compounds, including Ph, 4-NP, 2,4-DNP, 2-NP, 4-C-3-MP, 2,4-DCP, 2-M-4,6-DNP and 2,4,6-TCP. In order to obtain the best extraction efficiency, various extraction parameters, including the amount of sorbent, desorption solvent and its volume, vortex and centrifugation time for adsorption and desorption were optimized. The test solutions, containing analytes at 0.80 µg mL−1 for 4-C-3-MP, 2,4-DCP and 2,4,6-TCP and 0.50 µg mL−1 for the other five phenols, were used to perform the optimization experiments which were carried out in triplicate.
Effect of the Sorbent Amount
An appropriate sorbent amount is crucial for excellent extraction efficiency. The goal was to minimize it as much as possible, to ensure a real microextraction procedure. To study the influence of sorbent amount on adsorption efficiency, 10.00 mL of mixed standard solution of phenol compounds was added to different amounts of solid sorbent, then vortexed for 10 s. In the present work, the amount of NH2-MIL-53(Al) in the range of 10–120 mg was tested. After centrifugation for 10 min, the supernatant was filtered through a 0.45-μm membrane and the remaining analytes were analyzed by HPLC–PDA. The adsorption percentage (%Adsorption) of each analyte was calculated by the following equation:
where CA is the concentration of analyte adsorbed on the sorbent and C0 is the initial concentration of analyte.
The results in Fig. S2 (Supplementary Information) revealed that the adsorption increased with the increase of sorbent amounts from 10 to 30 mg and remained constant afterwards. No analytes except for Ph was detected after the adsorption experiment, indicating that the sorbent has good adsorption capability for the target analytes. Consequently, 30 mg of NH2-MIL-53(Al) was sufficient for effective extraction of the target analytes and was selected for further experiments.
The possible adsorption mechanism of phenol molecules on NH2-MIL-53(Al) is based on the π–π interaction between phenols and phenyl rings of aminoterephthalic acid ligand in the framework of NH2-MIL-53(Al). In addition, all of the studied phenols can enter the pores of NH2-MIL-53(Al) as their kinetic dynamic diameters (3.9–5.4 Å) [36] are smaller than the pores (7.3 Å × 7.7 Å) [37] of NH2-MIL-53(Al). Moreover, the hydrogen bonding can be formed between the nitro group of the nitrophenol as proton acceptor and amino groups as proton donors in the framework [24]. In case of Ph, the adsorption was unable to reach 100% because it contains only one hydroxyl group for forming hydrogen bonding and other noncovalent weak interactions with NH2-MIL-53(Al), leading to a less adsorption efficiency compared with other analytes [38].
Effect of the Adsorption Time
The adsorption time is one of the most important factors affecting the adsorption efficiency of analytes. To ensure that the analytes were adsorbed to a maximum extent, the vortex was selected for accelerating the adsorption of analytes onto the sorbent. The effect of vortex time was investigated in the range of 0–60 s using the sorbent amount of 30 mg and sample volume of 10.00 mL. As illustrated in Fig. S3 (Supplementary Information), the adsorption of 4-NP increased significantly in case of using vortex for 10 s when compared to the result obtained from the experiment without vortex process. The satisfactory results for all analytes could be obtained using vortex agitation for 10 s and further increase of vortex time did not contribute to any improvement of the %Adsorption. Therefore, the obtained results led to the selection of 10 s adsorption time using vortex agitation for further studies. At this point, the centrifugation after adsorption process was also optimized. Different centrifugation times were studied in the range of 1–13 min, but 1 min was found to be enough to settle down the sorbent and gave adequate extraction efficiency. Longer times did not improve any extraction efficiency (data not shown). Therefore, after adsorption process, the mixture was centrifuged for 1 min to separate the solid sorbent from the aqueous sample solution.
Effect of Desorption Solvent and Its Volume
The desorption solvent is also a critical variable for VA-D-µ-SPE technique. In this study, several solvents and solvent mixtures including methanol, acetonitrile, ethanol, methanol–acetic acid (9.5:0.5, v/v), acetonitrile–acetic acid (9.5:0.5, v/v) and ethanol–acetic acid (9.5:0.5, v/v) were evaluated as desorption solvent with vortex for 10 s for desorption of the analytes from the solid sorbent. The other experimental conditions were kept as follows: sample volume, 10.00 mL; sorbent amount, 30 mg; vortex time, 10 s; centrifugation after adsorption, 1 min at 5000 rpm; desorption solvent volume, 1000 μL; centrifugation after desorption, 10 min at 5000 rpm. The obtained results in Fig. S4 (Supplementary Information) show that high desorption efficiency was obtained using acetonitrile as desorption solvent. Moreover, the addition of acetic acid resulted in increasing of extraction recoveries for some analytes, i.e., Ph, 2,4-DNP, 2-NP and 2-M-4,6-DNP, due to its strongest dissolving ability to analytes. Thus, the mixture of acetonitrile–acetic acid (9.5:0.5) was chosen as desorption solvent for further studies.
The volume of the acetonitrile–acetic acid mixture was investigated in the range of 100–2000 μL. It should be noted that in this optimization process the solvent containing desorbed analytes was injected into the HPLC system without evaporation. As shown in Fig. S5 (Supplementary Information), the peak area of the analytes increased with increasing the volume of desorption solvent from 125–1500 μL and remained almost constant afterward. Therefore, 1500 μL of acetonitrile–acetic acid mixture was selected for desorption of the phenol compounds from NH2-MIL-53(Al) sorbent.
Effect of Desorption Time
The desorption time is another important factor that affects the extraction recovery of analyte. In this work, vortex was chosen to enhance the desorption of analytes from sorbent. The vortex desorption times were varied in the range of 0–60 s. The highest desorption of analytes were obtained when the vortex desorption time was at 10 s. After 10 s, the desorption remained constant (see Fig. S6, Supplementary Information). Hence, the vortex desorption time of 10 s was selected as optimum condition. At this point, the centrifugation time was also optimized. Different times in the range of 1–10 min were studied; however, 1 min was adequate to separate sorbent from solution. Longer times did not improve the extraction of analytes (data not shown).
Considering the results obtained from adsorption experiments, it could be observed that the NH2-MIL-53(Al) sorbent exhibited effectively quantitative adsorption for most phenol compounds (except for Ph). However, desorption of total amounts of adsorbed analytes from the sorbent could not be performed as expected. Only about 20% (for 2,4-DNP) to 65% (for 4-NP) were desorbed from the sorbent under the selected extraction condition. Anyway, the satisfactory sensitivity was achieved for determination of phenol compounds in real samples. In the present work, the sorbent was disposed after each determination for preventing the effect of carry over.
Analytical Features of Direct HPLC and the Proposed VA-D-µ-SPE-HPLC Procedure
Under the optimum HPLC condition as described in the experimental section, separation of eight phenols was achieved within 14 min, with the following order of elution: Ph (tR = 4.0 min), 4-NP (tR = 4.9 min), 2,4-DNP (tR = 7.0 min), 2-NP (tR = 8.1 min), 4-C-3-MP (tR = 9.5 min), 2,4-DCP (tR = 10.6 min), 2-M-4,6-DNP (tR = 11.3 min), and 2,4,6-TCP (tR = 13.8 min). Linearity was observed in the concentration range of 0.4000–50.000 µg mL−1 with the coefficients of determination (R2) greater than 0.9936. The limits of detection (LODs) and limits of quantification (LOQs) were considered as the concentrations obtaining a signal-to-noise ratio of 3 and 10, respectively. The LODs and LOQs ranged from 0.12–0.46 and 0.39–1.54 µg mL−1, respectively. The intra-day precision (n = 5) of the method, expressed as the relative standard deviations (RSDs) of the retention time and peak area of the phenols at 5 µg mL−1, was less than 0.33 and 4.36%, respectively. The inter-day experiments (n = 5 × 3) showed the RSDs of the retention time and peak area of less than 0.64 and 6.30%, respectively.
For preconcentration of phenol compounds, the optimum VA-D-µ-SPE condition using NH2-MIL-53(Al) as sorbent was applied before analysis by HPLC. This method exhibited a good linearity in the range of 0.0015–10.000 µg mL−1 with the R2 greater than 0.9929. The low LODs and LOQs were obtained in the range of 0.0004–0.0133 and 0.0013–0.0519 µg mL−1, respectively. Precisions in terms of intra- and inter-day experiments were investigated by replicate analyses of standard mixture of the analytes (0.05 µg mL−1 each) in a day (n = 5) and several days (n = 5 × 3), respectively. The intra-day RSDs of retention time and peak area were below 2.24 and 9.05%, respectively. For inter-day experiments, the RSD values were below 2.03 and 9.56% for retention time and peak area, respectively. The enrichment factors (EFs) for all analytes, which were obtained by comparing the concentrations before and after the VA-D-µ-SPE process, were in the range of 45 to 205. The analytical features of the proposed method are summarized in Table 1. Chromatograms of phenols obtained from the direct HPLC and the proposed VA-D-µ-SPE procedure are presented in Fig. 2.

Chromatograms of the phenols obtained by direct HPLC (1.0 µg mL−1) and concentrated by VA-D-μ-SPE method (0.05 µg mL−1). Detection at 280 nm. Peak assignment: 1, Ph; 2, 4-NP; 3, 2,4-DNP; 4, 2-NP; 5, 4-C-3-MP; 6, 2,4-DCP; 7, 2-M-4,5-DNP; 8, 2,4,6-TCP
Application to Real Samples
To evaluate the applicability of the developed VA-D-µ-SPE-HPLC for determination of phenol compounds in real sample matrices, 11 water samples, including lake water, treated waste waters, river water, seawater, tap water and three drinking waters were analyzed. No analyte residue was detected in the studied samples. In order to evaluate the accuracy, a recovery study was performed by spiking the samples with the analytes at the concentration levels of 0.10 and 0.15 µg mL−1 before analysis by the proposed method. As summarized in Table 2, the recoveries were all in the acceptable range of 72.3–111.4% (on average) with RSDs less than 10.4%. Fig. S7 (Supplementary Information) shows typical chromatograms of tap water sample with spiked phenols at various concentrations. Based on the results above, the proposed method gave good analytical performance for the analysis of target phenols in the studied water samples.
Comparison of the VA-D-µ-SPE to Other Methods
The developed VA-D-µ-SPE procedure using NH2-MIL-53(Al) as sorbent for determination of phenol compounds was compared to different SPE techniques, as summarized in Table 3. The sensitivity in term of LODs is comparable to that obtained from the MSPE method using molecular imprinted polymer [5]. However, the wide linear range for determining a variety of phenol compounds was obtained using the proposed procedure. For comparison with other MOF sorbents, the amount of sorbent, extraction conditions, linearity and LODs were considered. As can be seen in Table 3, the proposed NH2-MIL-53(Al) sorbent exhibits shorter extraction time and wider linearity range compared with those obtained from Fe3O4@SiO2-MOF-177 [36], Zn/Co7:1-MPC [39] and UiO-66-coated stainless steel fiber [40]. In addition, the developed method offers simple extraction process in short extraction time (less than 3 min) and low sample consumption, compared to those reported by other methods, in which can meet the requirements of the determination of a variety of phenolic compounds in various water matrices.
Molecular Docking
The binding free energies (∆Gbinding) obtained from molecular docking are listed in Table 4. Note that, a larger negative value of ∆Gbinding refers to higher analyte–MOFs interaction and adsorption ability. As a result, by substituting one nitro group the interaction was increased about 1.2 times compared to that of Ph. The increase of more than 1.3 times was obtained when substituting with two nitro groups, especially, when methyl group was included. This is due to not only the hydroxyl group, but also the substituted nitro groups can form hydrogen bonding with MOFs, leading to the larger binding ability (see Fig. 3b). Similarly, chloro substitutions provided the higher energy than − 6 kcal/mol (about 1.3 times) due to the dipole–dipole interaction between the chloro groups and polar hydrogen atoms of NH2-MIL-53(Al) taking place. In addition, the binding modes showed that the orientations of 4-C-3-MP, 2,4-DNP and 2-M-4,6-DNP molecular planes were parallel to the benzyl group of MOFs yielding the π–π interaction and thus enhancing the interaction. Parallel orientations were not observed for other compounds, as displayed in Fig. 3a (for other analytes, see also Fig. S8, Supporting Information). In summary, the calculated binding free energies are in good agreement with the adsorption ability observed experimentally.

Binding modes between MOFs and selected analytes exhibiting a orientation of the analytes in binding pocket and b hydrogen bonding (dashed line). The figures were generated by UCFS Chimera 1.11 program [39]
Conclusions
In this work, the metal–organic framework, NH2-MIL-53(Al) was successfully applied as sorbent for the VA-D-µ-SPE of eight phenols in different water samples. The developed approach exhibits good analytical performance and offers the simple operation process with time-saving and small sorbent amount usage. Furthermore, the docking calculations were investigated in terms of binding free energy and orientation of the analytes in the binding site of NH2-MIL-53(Al). The computational results were in good agreement with the experimental studies. The results proved that the NH2-MIL-53(Al) sorbent possesses great potential in the preconcentration of phenolic pollutants in trace levels.
References
Zhang PP, Shi ZG, Feng YQ (2011) Determination of phenols in environmental water samples by two-step liquid-phase microextraction coupled with high performance liquid chromatography. Talanta 85:2581–2586
Tang S, Lin XH, Li SFY, Lee HK (2014) In-syringe dispersive solid-phase extraction using dissolvable layered double oxide hollow spheres as sorbent followed by high-performance liquid chromatography for determination of 11 phenols in river water. J Chromatogr A 1373:31–39
Guan W, Han C, Wang X, Zou X, Pan J, Huo P, Li C (2012) Molecularly imprinted polymer surfaces as solid-phase extraction sorbents for the extraction of 2-nitrophenol and isomers from environmental water. J Sep Sci 35:490–497
Villar-Navarro M, Ramos-Payán M, Pérez-Bernal JL, Fernández-Torres R, Callejón-Mochón M, Bello-López MA (2012) Application of three phase hollow fiber based liquid phase microextraction (HF-LPME) for the simultaneous HPLC determination of phenol substituting compounds (alkyl-, chloro-, and nitrophenols). Talanta 99:55–61
Feng Q, Zhao L, Lin JM (2009) Molecularly imprinted polymer as micro-solid phase extraction combined with high performance liquid chromatography to determine phenolic compounds in environmental water samples. Anal Chim Acta 650:70–76
Zhou C, Tong S, Chang Y, Jia Q, Zhou W (2012) Ionic liquid based dispersive liquid–liquid microextraction with back-extraction coupled with capillary electrophoresis to determine phenolic compounds. Electrophoresis 33:1331–1338
Faraji H, Tehrani MS, Husain SW (2009) Pre-concentration of phenolic compounds in water samples by novel liquid–liquid microextraction and determination by gas chromatography–mass spectrometry. J Chromatogr A 1216:8569–8574
Bagheri H, Saraji M (2001) New polymeric sorbent for the solid-phase extraction of chlorophenols from water samples followed by gas chromatography–electron-capture detection. J Chromatogr A 910:87–93
Faraji H (2005) β-Cyclodextrin-bonded silica particles as the solid-phase extraction medium for the determination of phenol compounds in water samples followed by gas chromatography with flame ionization and mass spectrometry detection. J Chromatogr A 1087:283–288
Mousavi M, Noroozian E, Jalali-Heravi M, Mollahosseini A (2007) Optimization of solid-phase microextraction of volatile phenols in water by a polyaniline-coated Pt-fiber using experimental design. Anal Chim Acta 581:71–77
Montero L, Conradi S, Weiss H, Popp P (2005) Determination of phenols in lake and ground water samples by stir bar sorptive extraction–thermal desorption–gas chromatography–mass spectrometry. J Chromatogr A 1071:163–169
Zhao L, Lee HK (2001) Determination of phenols in water using liquid phase microextraction with back extraction combined with high-performance liquid chromatography. J Chromatogr A 931:95–105
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86:412–431
Dai X, Jia X, Zhao P, Wang T, Wang J, Huang P, He L, Hou X (2016) A combined experimental/computational study on metal-organic framework MIL-101(Cr) as a SPE sorbent for the determination of sulphonamides in environmental water samples coupling with UPLC-MS/MS. Talanta 154:581–588
Salisaeng P, Arnnok P, Patdhanagul N, Burakham R (2016) Vortex-assisted dispersive micro-solid phase extraction using CTAB modified Zeolite NaY sorbent coupled with HPLC for the determination of carbamate insecticides. J Agric Food Chem 64:2145–2152
Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM (2013) The chemistry and applications of metal-organic frameworks. Science 341:1230444
Boutin A, Couck S, Coudert F-X, Serra-Crespo P, Gascon J, Kapteijn F, Fuchs AH, Denayer JFM (2011) Thermodynamic analysis of the breathing of amino-functionalized MIL-53(Al) upon CO2 adsorption. Micropor Mesopor Mater 140:108–113
Zárate A, Peralt RA, Bayliss PA, Howie R, Sánchez-Serratos M, Carmona-Monroy P, Solis-Ibarra D, González-Zamora E, Ibarra IA (2016) CO2 capture under humid conditions in NH2-MIL-53(Al): the influence of the amine functional group. RSC Adv 6:9978–9983
Yusuf K, Aqel A, Alothman Z (2014) Metal-organic frameworks in chromatography. J Chromatogr A 1348:1–16
Serra-Crespo P, Berger R, Yang W, Gascon J, Kapteijn F (2015) Separation of CO2/CH4 mixtures over NH2-MIL-53-An experimental and modelling study. Chem Eng Sci 124:96–108
Serra-Crespo P, Ramos-Fernandez EV, Gascon J, Kapteijn F (2011) Synthesis and characterization of an amino functionalized MIL-101(Al): separation and catalytic properties. Chem Mater 23:2565–2572
Gascon J, Aktay U, Hernandez-Alonso MD, van Klink GPM, Kapteijn F (2009) Amino-based metal-organic frameworks as stable, highly active basic catalysts. J Catal 261:75–87
Lu T, Zhang L, Sun M, Deng D, Su Y, Lv Y (2016) Amino-functionalized metal-organic frameworks nanoplates-based energy transfer probe for highly selective fluorescence detection of free chlorine. Anal Chem 88:3413–3420
Liu B, Yang F, Zou Y, Peng Y (2014) Adsorption of phenol and p-nitrophenol from aqueous solutions on metal-organic frameworks: effect of hydrogen bonding. J Chem Eng Data 59:1476–1482
Hasan Z, Jhung SH (2015) Removal of hazardous organics from water using metal-organic frameworks (MOFs): plausible mechanisms for selective adsorptions. J Hazard Mater 283:329–339
Knichal JV, Gee WJ, Burrows AD, Raithby PR, Wilson CC (2015) A new small molecule gelator and 3D framework ligator of lead(II). Cryst Eng Comm 17:8139–8145
Sengupta S, Mondal R (2014) A novel gel-based approach to wastewater treatment—unique one-shot solution to potentially toxic metal and dye removal problems. J Mater Chem A 2:16373–16377
Rocío-Bautista P, González-Hernández P, Pino V, Pasán J, Afonso AM (2017) Metal-organic frameworks as novel sorbents in dispersive-based microextraction approaches. Trends Anal Chem 90:114–134
Ahnfeldt T, Gunzelmann D, Loiseau T, Hirsemann D, Senker J, Férey G, Stock N (2009) Synthesis and modification of a functionalized 3D open-framework structure with MIL-53 topology. Inorg Chem 48:3057–3064
Couck S, Denayer JFM, Baron GV, Rémy T, Gascon J, Kapteijn F (2009) An amine-functionalized MIL-53 metal-organic framework with large separation power for CO2 and CH4. J Am Chem Soc 131:6326–6327
Luo X, Li G, Hu Y (2017) In-tube solid-phase microextraction based on NH2-MIL-53(Al)-polymer monolithic column for online coupling with high-performance liquid chromatography for directly sensitive analysis of estrogens in human urine. Talanta 165:377–383
Chin JM, Chen EY, Menon AG, Tan HY, Hor ATS, Schreyer MK, Xu JW (2013) Tuning the aspect ratio of NH2-MIL-53(Al) microneedles and nanorods via coordination modulation. Cryst Eng Comm 15:654–657
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr. JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09 (Revision B.01), Gaussian, Inc., Wallingford
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 16:2785–2791
Seoane B, Téllez C, Coronas J, Staudt C (2013) NH2-MIL-53(Al) and NH2-MIL-101(Al) in sulfur-containing copolyimide mixed matrix membranes for gas separation. Sep Purif Technol 111:72–81
Wang GH, Lei YQ, Song HC (2014) Evaluation of Fe3O4@SiO2–MOF-177 as an advantageous adsorbent for magnetic solid-phase extraction of phenols in environmental water samples. Anal Methods 6:7842–7847
Loiseau T, Serre C, Huguenard C, Fink G, Taulelle F, Henry M, Bataille T, Fÿrey G (2004) A rationale for the large breathing of the porous aluminum terephthalate (MIL-53) upon hydration. Chem Eur J 10:1373–1382
Knichal JV, Gee WJ, Burrows AD, Raithby PR, Wilson CC (2015) Role of ethynyl-derived weak hydrogen-bond interactions in the supramolecular structures of 1D, 2D, and 3D coordination polymers containing 5-ethynyl-1,3-benzenedicarboxylate. Cryst Growth Des 15:465–474
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Li M, Wang J, Jiao C, Wang C, Wu Q, Wang Z (2016) Magnetic porous carbon derived from a Zn/Co bimetallic metal-organic framework as an adsorbent for the extraction of chlorophenols from water and honey tea samples. J Sep Sci 39:1884–1891
Shang H-B, Yang C-X, Yan X-P (2014) Metal-organic framework UiO-66 coated stainless steel fiber for solid-phase microextraction of phenols in water samples. J Chromatogr A 1357:165–171
Acknowledgements
The authors gratefully acknowledge financial support from the Royal Golden Jubilee (RGJ) Ph.D. program (Grant No. PHD/0050/2557). R. Burakham thanks the Thailand Research Fund (TRF) and Khon Kaen University for supporting the TRF Research Scholar (Grant No. RSA5980034). Partial supports from the Center for Innovation in Chemistry (PERCH-CIC), Office of the Higher Education Commission, Ministry of Education, Materials Chemistry Research Center, Khon Kaen University, and the TRF Distinguished Research Professor grant (Prof. Kate Grudpan) are also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Boontongto, T., Siriwong, K. & Burakham, R. Amine-Functionalized Metal–Organic Framework as a New Sorbent for Vortex-Assisted Dispersive Micro-Solid Phase Extraction of Phenol Residues in Water Samples Prior to HPLC Analysis: Experimental and Computational Studies. Chromatographia 81, 735–747 (2018). https://doi.org/10.1007/s10337-018-3498-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-018-3498-0