
Abstract
In this paper, a graphene-based magnetic nanocomposite (Fe3O4@G) was synthesized by covalent bonding. The presence of Fe3O4 was characterized by fourier transform infrared spectroscopy, X-ray diffraction, thermogravimetric analysis, scanning electron microscopy (SEM), and transmission electron microscopy. Subsequently, the Fe3O4@G was used as adsorbent for the extraction of eight organochlorine pesticides in orange juice samples prior to their gas chromatography mass spectrometry detection. Various experimental parameters affecting the extraction efficiencies, such as the amount of Fe3O4@G, extraction time, the pH of the sample solution and desorption conditions were investigated. Under optimized experimental conditions, good linearity existed in the range of 1.0–200.0 ng mL−1 for all the analytes with the correlation coefficients (R 2) ranging from 0.9904 to 0.9992. The limits of detection (S/N = 3) of the method for the compounds were between 0.01 and 0.05 ng mL−1. Good reproducibilities were obtained with the relative standard deviations (RSDs) below 9.6 %. The recoveries of the method were in the range between 73.8 and 105.4 % with RSDs less than 7.7 %.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Organochlorine pesticides (OCPs) such as hexachlorocyclohexanes (HCHs of α-, β-, δ-, and γ-isomers) and dichlorodiphenyltrichloroethane (DDT) have been used extensively to protect against a great variety of pests and diseases in the past few decades. It was estimated that the total worldwide production exceeded 10 million tons for HCHs [1]. HCHs and DDT were already banned in the 1970s in the United States due to their carcinogenicity, persistence in environment, long-distance transport, and bioaccumulation along the food chain [2]. Widespread consumption and low biodegradation of OCPs have nevertheless led to the ubiquitous presence of these toxic chemicals in the natural environment. Therefore, the trace analysis of OCPs residues in food samples is great importance for environmental control and human health protection. Nowadays, the determination of OCPs in vegetation and other matrices is often performed by gas chromatography (GC) with specific detectors, for example, electron capture detection (ECD) [3] and mass spectrometry (MS) [4]. These techniques offer the advantages of good sensitivity. The MS detector is widely used because of their selectivity and low detection limits [5]. In addition, the selected ion monitoring (SIM) mode allows reducing background noise. However, the sample matrices are complex, and the OCPs are in extremely low concentrations in food samples, which is inadequate for direct instrumental analysis. Hence, extraction and enrichment of OCPs in samples were needed prior to instrumental analysis. In recent years, magnetic solid-phase extraction (MSPE) as a new mode of SPE, which is based on the use of sorbents with paramagnetic features, has been widely applied for various samples [6–10]. The main advantage of MSPE is the increase in active contact area between the analytes and adsorbent. Thus, the extraction efficiency is high with less extraction time and the blocking problems, as occurs frequently in solid-phase extraction (SPE) cartridge or column, is avoided. For MSPE method, the application of magnetic sorption material with a high loading capacity and selective adsorption ability is critical.
Graphene, an atomically thin honeycomb lattice of carbon atoms, arouse great interest among scientists in various fields discovered shortly by Geim et al. in 2004 [11]. Due to its large specific surface area (2630 m2 g−1 from theoretical calculations based on single-layer graphene sheets) [12], chemical stability, flexibility and π-electron rich structure, etc., make a good candidate as a sorbent for sample pretreatment [13–16]. However, there are some problems for directly used graphene as adsorbents. First, irreversible aggregation may occur due to their high surface area during isolation from a well-dispersed solution by filtration or centrifugation. Second, graphene sheets may escape from SPE cartridge or column, especially under high pressure. Third, it is difficult to completely collect the miniscule graphene sheets from a homogeneous solution [17]. These problems may not only hinder effective sorption behaviors and reduce the sorption capacity, but also result into the SPE cartridge blocking. Introduction of Fe3O4 nanoparticles into graphene sheets can solve the above-mentioned problems and maintain the high adsorption performance of graphene sheets and the separation convenience of magnetic materials. Recently, a few graphene-based magnetic materials have been prepared for the preconcentration of some analytes, such as carbamate pesticides [18], small molecules [19], phthalate acid esters [9], sulfonamide antibiotics [20], triazine herbicides [21], proteins, and peptides [22]. However, most of the Fe3O4 nanoparticles were immobilized on graphene surface only by physical adsorption or electrostatic interaction, and they may be easily fall off from the graphene sheets during application [23].
In this work, the graphene-coated Fe3O4 nanoparticles (Fe3O4@G) were prepared by covalent bonding. The performance of Fe3O4@G as MSPE sorbent for the extraction of OCPs from orange juice samples prior to GC–MS analysis was first demonstrated. Several experimental parameters that could affect the extraction efficiencies such as the amount of Fe3O4@G, extraction time, sample solution pH, and desorption conditions were evaluated. The results indicated that the developed method was simple and effective for the determination of the OCPs in the juice with good sensitivity and reproducibility.
Experimental
Reagents and Materials
Graphite flakes (99.95 %, particle size ≤30 µm) were purchased from Alfa Aesar (Heysham, England). TEOS and APTES were purchased from Chengdu apotheker chemical reagent co., LTD (Chengdu, China). Potassium permanganate (KMnO4), sulphuric acid (H2SO4), Ferric chloride (FeCl3·6H2O), ferrous sulfate (FeSO4·7H2O), aqueous ammonia, N,N-dimethylformamide (DMF), ethyl alcohol absolute were supplied by Beijing Chemical Works (Beijing, China). All of the organic reagents employed were analytical purity. Deionized water was used throughout experiments. Pesticide standards of OCPs including α-, β-, γ-, and δ-HCHs, p,p′-DDT, o,p′-DDT, p,p′-DDE, and p,p′-DDD were purchased from the Institute for Environmental Reference Materials of Ministry of Environmental Protection (Beijing, China), which were prepared in methanol containing a mixture of eight OCPs at the concentration of 100 µg mL−1. A series of standard solutions were prepared by the appropriate dilution of the stock solutions and stored at 4 °C in a freezer. All the orange juice samples were purchased from local supermarkets in Changchun (Changchun, China) and stored at 4 °C.
Instruments and Analytical Conditions
Infrared absorption spectra were obtained with a Varian 800 FT-IR spectrometer (Varian, Palo Alto, USA). XRD spectra were carried out using an XPert Powder X-ray diffractometer (Panalytical, Almelo, Holland). TGA was characterized by a simultaneous thermal analyzer STA 409 PC Luxx (Netzsch, Germany). SEM images were recorded on a S-4800 instrument (Hitachi, Japan). TEM images were obtained with a H-600 transmission electron microscope (Hitachi, Japan).
A 7890A GC and a quadrupole MS 5975C (Agilent, Palo Alto, CA) with a 30 m × 0.25 mm, 0.25 µm film DB-17 bonded-phase fused silica capillary column (J&W Scientific, Inc., Folsom, CA) was used. Helium carrier gas was set at a flow rate of 1.0 mL min−1, and separation of the eight OCPs was carried out under a temperature programmed as follows: injector temperature at 200 °C, column temperature with an initial temperature of 90 °C (1-min hold) and ramped to 200 °C at a rate of 50 °C min−1 for 6 min, finally increased to 280 °C at the rate of 10 °C min−1 and held for 5 min. The ions source was set at 230 °C, the mass range was from m/z 100 to m/z 500, and the ionization energy was under electron ionization mode at 70 eV. Aliquots of 1 µL were injected into the GC–MS system under splitless mode, and the analytes were, respectively, monitored under the selected ion monitoring mode at multiple mass channels. Retention times, selected quantification and identification ions of the eight OCPs are shown in Electronic Supplementary Material Table S1.
Preparation of Fe3O4@G Nanocomposites
Fe3O4 nanoparticles were prepared by microwave synthesis [24]. The amino functionalized Fe3O4 nanoparticles were synthesized according to the literature [25]. Graphite oxide (GO) was prepared by a modified Hummers method [26]. GO sheets, owning a considerable amount of epoxide and hydroxyl functional groups on both surfaces of each sheet and carboxyl groups mostly at the sheet edges, are strongly hydrophilic and can form well-dispersed aqueous colloids. Generally, large amounts of graphene are most easily produced via the reduction of GO, which offers tremendous opportunities for chemical bonded graphene MSPE coating preparation. According to that Fe3O4@GO was fabricated via the carboxy groups of GO were linked to the amino groups of amino functionalized Fe3O4 in aqueous solutions. The Fe3O4@G was prepared of Fe3O4 nanoparticles encapsulated GO (Fe3O4@GO) by a hydrothermal reaction. Typically, 150 mg GO in 300 mL of water was ultrasonicated for 3 h, then 150 mg amino functionalized Fe3O4 dispersion was added into the homogenous suspension and mixture was ultrasonication for 30 min. After stirring for 2 h at 80 °C, the mixture was transferred into a 400 mL Teflon-lined stainless-steel autoclave and reacted at 180 °C for 3 h. The precipitate was collected by a magnet and washed with DMF and water. Finally, the resulted product was dried under vacuum at 60 °C.
Sample Preparation
The samples were diluted with deionized water (1:1, v/v) and then centrifuged at 4000 rpm for 5 min. The supernatant was collected and filtered through a 0.45 µm membrane to eliminate particulate matters before analysis. The 1:1 diluted orange juice samples with water are named ‘diluted juice sample’ hereafter.
MSPE Procedure
The MSPE procedure is shown in Fig. 1. Typically, 40 mL of diluted juice sample with desired initial concentration of OCPs and 20 mg of Fe3O4@G were added in a 50 mL centrifuge tube and then the tube was shaken on a platform shaker under ambient temperature. After 10 min, an external permanent magnet was placed beside the centrifuge tube to hold the Fe3O4@G which had already extracted the analytes. The supernatant was discarded. Subsequently, the residual supernatant and Fe3O4@G were totally transferred to a 2 mL centrifuge tube. The adsorbent was gathered again by placing a magnet to the outside of tube wall to completely remove the residual solution. After being washed with deionized water to remove impurities that may exist, the preconcentrated target analytes were eluted from the isolated Fe3O4@G with 0.5 mL of mixed solvents of acetone and hexane (1:1, v/v) by vigorously vortexing for 1 min. This desorption procedure was operated another two times. The supernatant solutions were combined together, and then evaporated to dryness under a gentle stream of nitrogen gas. The residue was dissolved in 50 µL of methanol, and 1 µL was injected into the GC–MS to analyze.

The overall synthetic procedure of Fe3O4@G nanocomposites and the MSPE procedure when Fe3O4@ G was used as sorbent
Optimization of Adsorption and Desorption Conditions
To investigate the extraction performance, extraction recovery (R) was used. R is expressed as follows:
where C is the analyte concentration (ng mL−1) in the reconstituted solvent, C 0 is the initial concentration of analyte in water sample. V and V 0 are the volumes of the reconstituted solvent and water sample, respectively.
In the experiment of optimization of adsorption conditions, the adsorbent was loaded 40 mL diluted juice sample solution spiked with 2 µg of each pesticides, when methanol (3 mL, three times) was used as elution solvent. To evaluate the influence of sample pH on the extraction recoveries, the sample solution was added hydrochloric acid or sodium hydroxide to adjust the pH in range from 2.0 to 12.0. When evaluate the type of the elution, the adsorbent was loaded 40 mL diluted sample solution with 2 µg spiked of each analyte, then eluted with 3 mL (each time) of elution solvent three times. To investigate the reusability of Fe3O4@G, the adsorbent was washed with 3 mL of acetone twice and then with 3 mL water after desorption.
Validation of the MSPE-GC–MS Method
The linearity was studied in the range 1.0–200.0 ng mL−1, when a series of working standard samples were prepared by different concentration spiked OCPs-free juice samples for the establishment of the matrix-matched standard calibration curves. The limit of detection (LOD) and limit of quantification (LOQ) were estimated by five replicate analyses of the calibration solution with known concentration. The intra-day and inter-day precisions were investigated by the extraction and determination of the analytes from the spiked juice samples at 5.0 ng mL−1 of each pesticides in the same day and on three consecutive days. The relative standard deviation (RSD) was calculated on the basis of the obtained peak areas. Recovery was also investigated by spiking the analytes into the samples at concentrations of 5.0 and 20.0 ng mL−1. Triplicate measurements were performed by MSPE–GC–MS.
Results and Discussion
Characterization of the Fe3O4@G Nanocomposites
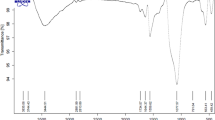
GO reduction during hydrothermal treatment was confirmed by FT-IR spectra. Figure 2a shows the FT-IR spectra of GO, G and Fe3O4@G. In GO, we observed the absorption bands at 3410, 1735, 1597, 1064 cm−1, which are attributed to the stretching vibrations of O–H, C=O, aromatic C=C, and alkoxy C–O. The IR spectrum of composite product confirmed the reduction of GO sheets because the absorption at 1735 cm−1 (C=O stretching vibrations) was decreased very much in intensity and most bands that are related with the oxygen-functional groups almost vanished. The absorption band that appears at 1597 cm−1 may be ascribed to the skeletal vibration of the graphene sheets. A new absorption band at 579 cm−1 can be attributed to lattice absorption of iron oxide [27], indicating that Fe3O4 was successfully grafted on G.

a FT-IR spectra and b XRD pattern of GO, G and Fe3O4@G nanocomposites; c TGA curve of the Fe3O4@G nanocomposites
To obtain the phase and crystal structure information about products, XRD measurements were conducted. Figure 2b illustrates the XRD pattern of graphite flakes, GO, G and Fe3O4@G with the corresponding 2θ values. After the oxidation of graphite flakes, reflection peak shifts to the lower angle at 2θ = 10.0° from the 2θ = 26.6° indicates the successful oxidation of graphite. In the XRD pattern of Fe3O4@G, seven diffraction peaks are observed at 2θ = 30.2°, 35.5°, 43.2°, 53.5°, 57.2°, 62.6°, 74.1° corresponding to the characteristic Fe3O4 planes of (2 2 0), (3 1 1), (4 0 0), (4 2 2), (5 1 1), (4 4 0), and (5 3 3), respectively. All the diffraction peaks can be perfectly indexed to the standard value of the Fe3O4 (JCPDS card No. 19-0629) phase, and no characteristic diffraction peak of GO was found after reduced by hydrothermal treatment, which is in agreement with the GO being reduced to G during the synthesis process. According to the spectra of G and Fe3O4@G, without extra diffraction peaks related to carbon, it can be concluded that the G sheets were completely exfoliated due to the loading of Fe3O4 nanoparticles on their surfaces [28]. These results suggested the presence of Fe3O4 nanoparticles on G sheets, which is consistent with the FT-IR results. To determine the mass content of G in the Fe3O4@G, TGA was performed in air at the temperature range from room temperature to 800 °C at a rate of 10 °C min−1. TGA curve of the Fe3O4@G is shown in Fig. 2c. A weight loss from room temperature to 250 °C which may be attributed to the desorption of surface bound water. An abrupt weight loss that occurs from 250 to 700 °C could be caused by the removal of oxygen-functional groups and the decomposition of carbon framework from the nanocomposites [29]. After 700 °C, there is no further mass loss, indicating the complete removal of graphene. According to the TGA curve, the content of G is about 34.71 wt%, which is calculated from the weight loss of 250–700 °C.
The morphology of Fe3O4@G was investigated by SEM and TEM. Figure 3 illustrates the SEM and TEM images of the Fe3O4@G. After a long time of sonication with ethanol during the preparation of the specimen, the SEM and TEM images were displayed that Fe3O4 nanoparticles deposited onto the surface of G sheets and no Fe3O4 nanoparticles falling off from G sheets, suggesting the strong interaction between Fe3O4 nanoparticles and G sheets. As observed from Fig. 3a, the G sheets look like transparent veil decorating Fe3O4 nanoparticles, implying that the G sheets are quite thin and had been exfoliated. In addition, the Fe3O4 nanoparticles are nearly spherical in shape and with a typical size of about 60 nm.

a SEM image and b TEM image of Fe3O4@G nanocomposites
Optimization of Adsorption and Desorption Conditions
To obtain the maximal extraction efficiency, several parameters including the amount of the magnetic graphene, extraction time, pH value of the sample, as well as the type and the volume of eluent solvent were investigated and optimized in this work.
Effect of the Amount of Fe3O4@G
The adsorbent amount had a significant effect on extraction efficiency. In this work, the amounts of Fe3O4@G in the range of 5–30 mg were investigated. According to the results shown in Fig. 4a, the extraction recovery increased as the amount of adsorbent increased. When the amount increased to 20 mg, the curves turned out to be flat, and there was no distinct increase to extraction recovery, indicating that 20 mg of sorbent was sufficient to extract OCPs from the sample solution. According to the result, 20 mg of Fe3O4@G was employed in the following studies.

Effect of extraction conditions on MSPE efficiency of the eight pesticides. a Effect of the amount of Fe3O4@G; b effect of extraction time; c effect of the sample solution pH; d effect of the desorption solvent
Effect of Extraction Time
In MSPE procedure, extraction time is an important parameter which can influence the efficiency of extraction. In this study, different extraction time (5, 8, 10, 15, and 30 min) was studied. As shown in Fig. 4b, the extraction recoveries were increased with increased extraction time from 5 to 10 min, and then remained almost constant after 10 min. Hence, the extraction time of 10 min was selected.
Effect of pH of the Sample
A suitable pH possibly could improve the extraction efficiency of the specific analyte on the adsorbent. Figure 4c shows that the extraction recovery of the analytes remained almost constant at pHs between 4.0 and 8.0; when the pH was lower than 2.0 or higher than 8.0, the extraction recovery was decreased. This was probably due to the pH either affecting the surface charge of the adsorbent or the physicochemical properties of the analyte. The pH of the diluted juice sample solutions was normally at about 4–5, thus there is no need to adjust the pH of the sample solution before extraction.
Type and Volume of Elution Solvent Selection
Selection of the kind of elution solvent is quite importance for the extraction efficiency of the analytes. In the current work, we selected methanol, acetone, hexane and acetone-hexane (1:1, v/v) as eluting solvent, and then compared their eluting efficiencies. According to the results shown in Fig. 4d methanol had poor eluting capability compared with the other elution solvents. Acetone improved the extraction recoveries of p,p′-DDE, p,p′-DDD, o,p′-DDT, and p,p′-DDT, but it had poor eluting ability for the HCHs. Hexane showed a good eluting ability, but it was only 90.2 and 86.7 % for o,p′-DDT and p,p′-DDT. When using acetone-hexane (1:1, v/v) as the eluting solvent, the highest extraction recoveries in the range from 98.7 to 103.5 % were obtained. Thus, acetone-hexane (1:1, v/v) was chosen as elution solvent. The volume of elution solvent is also a key factor to obtain reliable and reproducible analytical results. In the present study, the influence of the volume of elution solvent on the extraction recoveries was studied. It was found that the extraction with 0.5 mL acetone-hexane (1:1, v/v) three times could completely elute the analytes from the adsorbent.
Reusability of the Adsorbent
The adsorbent after processed was reused for the next MSPE procedure according to the section “MSPE Procedure”. In such a way, no carry-over of the analytes was detected on the adsorbent. The matrix of orange juice is extremely complex and contains varieties of chemical substances, especially pigments. After the extraction by Fe3O4@G, the concentrated elution was almost without color through visual observation. This phenomenon provides a clear proof that Fe3O4@G has the ability for the effective clean-up of the pigment, which is the main interference for analysis. The results showed that the magnetic adsorbent can be reused at least 15 times without significant loss of the extraction recoveries (>72.3 %) of OCPs. The fact indicates that Fe3O4@G has great potential for use in sample preparation for complex samples.
Validation of the MSPE-GC–MS Method
To evaluate the performance of the developed MSPE coupled with the GC–MS method for the analysis of the OCPs in orange juices, several important parameters including linearity, correlation coefficients (R 2), limits of detection (LODs), limits of quantification (LOQs), reproducibilities were determined under the above optimized experimental conditions and the results are summarized in Table 1. The calibration curve of each OCP was constructed by plotting the peak areas (y) versus the corresponding concentration of the analytes (x). As can be seen, good linearities were observed in the range of 1.0–200.0 ng mL−1 for all the OCPs with the correlation coefficients (R 2) ranging from 0.9904 to 0.9992. Based on a signal to noise ratio (S/N) of 3 and 10, the LODs and LOQs for the compounds were estimated to be in range from 0.01 to 0.05 ng mL−1 and from 0.06 to 0.15 ng mL−1, respectively. The within laboratory reproducibilities of the developed MSPE method were evaluated in terms of intra-day and inter-day precisions. The results, expressed as the relative standard deviation (RSD) of peak areas, are presented in Table 1. As can be observed, an acceptable precision was obtained with intraday RSD values below 7.7 % and interday RSD values within 9.6 %. Thereby, the proposed method is sensitive and repeatable.
Table 2 compares the performance of the proposed method in this study and other previously reported methods. The results show that the MSPE method has a shorter extraction time, wider linearity and a comparable reproducibility in contrast with the methods mentioned above. In addition, this method has lower LODs compared with the other method. These results confirmed that this method is faster, more sensitive and efficient.
Application of the Method to Orange Juice Samples
Under the optimal conditions, the current method was applied to the determination of the OCPs in three commercially available orange juice samples from a local market. The results show that no residues of the target OCPs were found in the samples. To validate the accuracy of the method, the recoveries of the method for the OCPs were investigated by spiking the analytes into the samples at concentrations of 5.0 and 20.0 ng mL−1, respectively. The results are given in Table 3, showing that the recoveries of the OCPs were in the range from 73.8 to 105.4 % with RSDs between 3.3 and 7.7 %. The data revealed that the new method was suitable for the analysis of the target analytes in real samples. Figure 5 shows typical chromatograms of the extracted analytes from orange juice sample before and after being spiked with each of the OCPs at 5 ng mL−1.

The total ion chromatograms obtained from unspiked and spiked sample. Peak identification: (1) α-HCH, (2) β-HCH, (3) γ-HCH, (4) δ-HCH, (5) p,p’-DDE, (6) o,p’-DDT, (7) p,p’-DDD, (8) p,p’-DDT
Conclusions
In summary, the Fe3O4 nanoparticles were grafted onto the surface of graphene by a chemical bonding method. The prepared Fe3O4@G was used as MSPE adsorbent for the enrichment of OCPs from real samples followed by GC–MS. We demonstrated that our Fe3O4@G had great potential as an effective absorbent for enriching the OCPs in fruit juice due to its high absorption capacity, convenient magnetic separation and the more complex sample matrices, and it would have a significant application potential for the enrichment of other environmental pollutants.
References
Li YF (1999) Sci Total Environ 232:121–158
Nollet LML, Rathore HS (2010) Handbook of pesticides: methods of pesticide residues analysis. CRC Press, Taylor Francis Group, New York
Domingues V, Cabral M, Alves A, Delerue-Matos C (2009) Anal Lett 42:706–726
Guan H, Brewer WE, Garris ST, Morgan SL (2010) J Chromatogr A 1217:1867–1874
Zrostlikova J, Lehotay SJ, Hajslova J (2002) J Sep Sci 25:527–537
Sukhanova II, Dikunets MA, Viryus ED, Rodchenkov GM (2011) J Anal Chem 66:807–814
Heidari H, Razmi H (2012) Talanta 99:13–21
Xie J, Liu TS, Song GX, Hu YM, Deng CH (2013) Chromatographia 76:535–540
Ye Q, Liu L, Chen Z, Hong L (2014) J Chromatogr A 1329:24–29
Wang L, Zang X, Chang Q, Wang C, Wang Z (2014) Anal Methods 6:253–260
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Science 306:666–669
Stoller MD, Park SJ, Zhu YW, An JH, Ruoff RS (2008) Nano Lett 8:3498–3502
Liu Q, Shi J, Zeng L, Wang T, Cai Y, Jiang GJ (2011) Chromatogr A 1218:197–204
Huang KJ, Yu S, Li J, Wu ZW, Wei CY (2012) Microchim Acta 176:327–335
Guan WB, Li ZN, Zhang HY, Hong HJ, Rebeyev N, Ye Y, Ma YQ (2013) J Chromatogr A 1286:1–8
Han Q, Wang Z, Xia J, Zhang X, Wang H, Ding M (2014) J Sep Sci 37:99–105
Liu Q, Shi JB, Sun JT, Wang T, Zeng LX, Jiang GB (2011) Angew Chem Int Ed 50:5913–5917
Wu Q, Zhao G, Feng C, Wang C, Wang Z (2011) J Chromatogr A 1218:7936–7942
Shi C, Meng J, Deng C (2012) Chem Commun 48:2418–2420
Luo YB, Shi ZG, Gao QA, Feng YQ (2011) J Chromatogr A 1218:1353–1358
Zhao G, Song S, Wang C, Wu Q, Wang Z (2011) Anal Chim Acta 708:155–159
Liu Q, Shi JB, Cheng MT, Li GL, Cao D, Jiang GB (2012) Chem Commun 48:1874–1876
He F, Fan J, Ma D, Zhang L, Leung C, Chan HL (2010) Carbon 48:3139–3144
Hong RY, Pan TT, Li HZ (2006) J Magn Magn Mater 303:60–68
Hu Y, Huang Z, Liao J, Li G (2013) Anal Chem 85:6885–6893
Tung VC, Chen LM, Allen MJ, Wassei JK, Nelson K, Kaner RB, Yang Y (2009) Nano Lett 9:1949–1955
Yan S, Qi TT, Chen DW, Lia Z, Li XJ, Pan SY (2014) J Chromatogr A 1347:30–38
Luo YB, Zhu GT, Li XS, Yuan BF, Feng YQ (2013) J ChromatogrA 1299:10–17
Bosch-Navarro C, Coronado E, Martí-Gastaldo C, Sánchez-Royo JF, Gòmez MG (2012) Nanoscale 4:3977–3982
Russo MV (2000) Chromatographia 51:71–76
Zhu P, Miao H, Du J, Zou J, Zhang G, Zhao Y, Wu Y (2014) J Agric Food Chem 62:7092–7100
Famiglini G, Palma P, Pierini E, Trufelli H, Cappiello A (2008) Anal Chem 80:3445–3449
Chan S, Kong MF, Wong YC, Wong SK, Sin Della WM (2007) J Agric Food Chem 55:3339–3345
Numata VM, Yarita T, Aoyagi Y, Takatsu A (2003) Anal Chem 75:1450–1457
Zhou T, Xiao X, Li G (2012) Anal Chem 84:420–427
Fernandes VC, Domingues VF, Mateus N, Delerue-Matos C (2011) J Agric Food Chem 59:7582–7591
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standard
This article does not contain any animal or human studies performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sun, T., Yang, J., Li, L. et al. Preparation of Graphene Sheets with Covalently Bonded Fe3O4 for Magnetic Solid-Phase Extraction Applied to Organochlorine Pesticides in Orange Juice. Chromatographia 79, 345–353 (2016). https://doi.org/10.1007/s10337-016-3024-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-016-3024-1