
Abstract
Hollow fiber-protected magnetized solvent-bar liquid-phase microextraction of sulfonylurea herbicides, including nicosulfuron, bensulfuron-methyl, pyrazosulfuron and chlorimuron-ethyl, in pears was developed. The analytes were determined by high-performance liquid chromatography. This present method has the advantages of stir bar sorptive extraction and hollow fiber-protected solvent-bar microextraction, that permits active magnetic stirring, extraction and preenrichment in a single device simultaneously. The magnetized solvent-bar was easily retrieved after extraction with a magnet, so the extract is conveniently collected. Experimental parameters, including type of extraction solvent, the number of magnetized solvent-bars, extraction temperature and time, stirring speed, pH of the sample solution, and the salting-out effect, were investigated and optimized. Under optimal experimental conditions, the calibration curve showed good linearity (r ≥ 0.9998). The limits of detection and quantification were in the range of 7.15–8.26 ng/g and 24.99–27.52 ng/g, respectively. The recoveries were in the range of 80.08–105.56 %, and the relative standard deviations were lower than 6.82 %. The results showed that the present method was a satisfactory method for the determination of sulfonylurea herbicides in pears.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sulfonylurea herbicides (SUHs) are composed of a sulphonyl structure linked to a urea group. The first commercialized sulfonylurea herbicide, sulfometuron-methyl, was registered by DuPont Agricultural Products in 1982 [1]. Over the years, SUHs were popularly used in the world due to their low application rates, high herbicidal activity, and low toxicity to mammals [2]. However, because of high mobility and increasing usage of SUHs, the residues of SUHs were detectable in the environment samples. The public and regulatory authorities felt some concern about potential adverse environmental impacts of SUHs, because SUHs could stop cell division in plants and lead to yield reduction in sensitive crops [3]. The maximum residue limit (MRL) of SUHs in the environment or agricultural products were set by many countries [4]. In the USA, the MRL of SUHs was 0.05 mg/kg in rice. In Japan, the MRL of imazosulfuron, bensulfuron methyl, and azimsulfuron was set at 0.1 mg/kg in rice. MRL of foramsulfuron and flazasulfuron was 0.01 mg/kg for litchi fruit in the European Union. In recent years, significant progress has been made towards the development of methods for the determination of sulfonylurea herbicides in environmental samples, such as water, soil, and plant tissues. However, most of the methods reported were time consuming and complicated pretreatment steps to obtain effective isolation of analyte from the samples were required [5]. Therefore, it is necessary to develop a more sensitive and faster method for determination of sulfonylurea herbicide residues in foods and environmental matrices.
Various methods have been developed for the determination of SUH residues, such as capillary electrophoresis (CE) [6], high-performance liquid chromatography (HPLC) [7, 8], liquid chromatography-tandem mass spectrometry (LC-MS/MS) [9] and gas chromatography (GC) [10]. SUHs are polar compounds with low volatility and thermal instability. HPLC is a good method for determining these polar and thermally labile herbicides, and the conventional ultraviolet (UV) detector has been extensively used.
Sample preparation is an important step in the whole analytical process. For determination of SUHs, several sample preparation methods have been developed, including supercritical fluid extraction (SFE) [11, 12], solid-phase extraction (SPE) [13, 14], molecularly imprinted SPE [7, 15], microwave-assisted solvent extraction (MASE) [16] and dispersive liquid–liquid microextraction (DLLME) [8].
Since 1999, hollow fiber liquid-phase microextraction (HF-LPME) introduced by Pedersen–Bjergaard and Rasmussen [17], has been widely applied as an alternative to the conventional method. In this method, the analytes are extracted from aqueous samples through a thin layer of organic solvent (several microlitres) immobilized within the pores of a porous hollow fiber, and transferred into an acceptor solution inside the hollow of the hollow fiber [18]. This method has advantages of preconcentration, sample cleanup, and solvent saving, and the required equipment is simple and inexpensive [19]. In addition, the small pore size prevents large molecules and particles present in the donor solution from entering the accepting phase and, at the same time, most macromolecules do not enter the hollow fiber because they are not soluble in the organic phase present in the pores in the extraction conditions, thus, yielding very clean extracts [20]. Several reviews on hollow fiber-based LPME have been reported [21–24]. Another alternative microextraction method, a variation of HF-LPME that involves a free-moving solvent-filled HF, solvent-bar microextraction (SBME) was proposed by Jiang and Lee [25]. The free movement of the solvent-bar in the aqueous sample solution greatly improves the transfer rate of analytes from the aqueous sample to the extraction solvent. Compared with SPE, when the SBME was applied, the extraction capacity is higher, the enrichment factor is higher and the surface area is larger [26, 27].
In this paper, a new hollow fiber-protected magnetized solvent-bar liquid-phase microextraction (HF-MSBLPME) was first applied for the extraction of four sulfonylurea herbicides from pears. This method combined the advantages of stir bar sorptive extraction and hollow fiber-protected solvent bar microextration. The stainless steel wire was inserted into the hollow of the hollow fiber to make the magnetized solvent-bar [28]. The magnetized solvent-bar permitted active magnetic stirring, extraction and preenrichment in a single step, and was easily retrieved after extraction with a magnet, so the extract is conveniently collected.
Experimental
Chemicals and Reagents
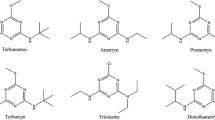
The standards, nicosulfuron, bensulfuron-methyl, pyrazosulfuron and chlorimuron-ethyl were obtained from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). The molecular structures of the analytes are shown in Fig. 1. The standard stock solutions of the compounds were prepared by dissolving the analytes in acetonitrile, and stored at 4 °C. Chromatographic grade acetonitrile and methanol were purchased from Fisher Corporation (Pittsburgh, PA, USA). All other analytical reagents were purchased from Beijing Chemical Factory (Beijing, China). Pure water was obtained with a Milli-Q water system (Millipore, Billerica, MA, USA). Q 3/2 accurel polypropylene hollow fiber (600 μm inner diameter, 200 μm wall thickness and 0.2 μm pore size) was purchased from Membrana GmbH (Wuppertal, Germany). The stainless steel wire (505 μm outer diameter) was inserted into the hollow of the hollow fiber.

Molecular structures of nicosulfuron, bensulfuron-methyl, pyrazosulfuron, and chlorimuron-ethyl
Instrument and Chromatographic Conditions
A Shimadzu LC-20A HPLC system (Shimadzu, Kyoto, Japan) equipped with a SPD-20A ultraviolet detector was used. Chromatographic separation was performed on a XDB-C18 column (150 mm × 4.6 mm I.D., 5 μm) (Agilent, Palo Alto, CA, USA). A KQ-100DE ultrasonic generator (Kunshan, Jiangsu, China) was used.
The mobile phase consisted of 0.1 % formic acid (A) and acetonitrile (B). The gradient condition was as follows: 0–3 min, 40–50 % B; 3–7 min, 50–55 % B; 7–9 min, 55–55 % B; 9–13 min, 55–50 % B; 13–16 min, 50–40 % B; and 16–20 min, 40–40 % B. The flow rate of the mobile phase was kept at 0.60 mL/min. The injection volume of the sample solution was 10 μL, and temperature of the column was controlled at 30 °C. The detection wavelength was set at 240 nm.
Sample Preparation
Fresh pear samples (Samples 1–4) were purchased from a local supermarket (Changchun, China). The present extraction procedure is not directly applicable to pear samples per se, and the samples need to be homogenized first. Thus, some form of pretreatment is necessary. The pear samples were homogenized with a food processor. The homogenized pear samples were weighed and spiked with working standard solutions. To ensure the standard solution to be well distributed in the sample, the standard solution was spiked in the homogenized sample, and the spiked samples were stored for 12 h in a dark place at room temperature, so the spiked samples can be homogenized better. Sample 1 was used in the optimization of experimental conditions. All experiments were performed in triplicate.
Hollow Fiber-Protected Magnetized Solvent-Bar Liquid-Phase Microextraction
The hollow fiber and the stainless steel wire were manually cut into 1.2 cm pieces, washed with acetone in a ultrasonic bath to remove impurities and dried in the air. The stainless steel wire was inserted into the hollow of the hollow fiber, by means of which the magnetized solvent-bar can stir in the magnetic field. Then the resulting hollow fiber was immersed in chloroform for 1 min in order to impregnate pores of the fiber wall, and rinsed with water in order to remove the extra chloroform from the surface of the fiber. The prepared hollow fiber was referred to as a magnetized solvent-bar.
Then 1.00 g of pear sample was weighed accurately and placed in a 20.00 mL extraction vessel. Then 5.00 mL of deionized water were added into the vessel and the pH of the sample solution was adjusted with 0.10 mol L−1 HCl to 3.0. Then the eight magnetized solvent-bars were placed into the sample solution. The vessel was placed on a magnetic stirrer and the stirring extraction was performed for 20 min at the extraction temperature of 30 °C and a stirring rate of 700 rpm. After extraction, the magnetized solvent-bars were separated rapidly from the solution with an external magnet. Then the analytes on the magnetized solvent-bars were desorbed with 500 μL of acetonitrile in ultrasonic bath for 5 min. The resulting solution was dried under nitrogen steam and the residue was dissolved in 100 μL acetonitrile. The resulting solution was filtered through a 0.22 μm membrane filter and then directly analyzed by HPLC.
Results and Discussion
Optimization of HF-MSBLPME
In HF-MSBLPME, many conditions can affect the extraction efficiency, such as the type of extraction solvent, the number of magnetized solvent-bars, extraction time and temperature, stirring speed, the pH of the sample solution and salt concentration. In this work, the effect of the conditions on the extraction recoveries was studied.
Type of Extraction Solvent
In HF-LPME, the selection of an appropriate extraction solvent is of major importance to achieve efficient extraction. There are some considerations in choosing extraction solvent, such as high extraction capability of analytes, immiscibility with water, low volatility, compatibility with HF, and less interference with the chromatographic analysis of the target analytes. In order to select a suitable extraction solvent, five kinds of organic solvents, including dichloromethane, chloroform, n-hexane, cyclohexane, and toluene were investigated. The recoveries obtained with the extraction solvents are shown in Fig. 2. The results show that the recoveries obtained with chloroform are higher than those obtained with other solvents. This may be due to the principle of extraction of ‘like dissolves like’. Therefore, chloroform was chosen as the extraction solvent in this investigation.

Effect of type of extraction solvent. Number of magnetized solvent-bars, 8; extraction time, 20 min; extraction temperature, 30 °C; stirring speed, 700 rpm; NaCl concentration, 10 %; pH of sample solution, 3.0
The Number of Magnetized Solvent-Bars
In order to evaluate the influence of extraction solvent volume on recoveries, the effect of the number of magnetized solvent-bars was studied, because the pores of hollow fiber were impregnated with extraction solvent. Figure 3 shows the effect of the number of magnetized solvent-bars on the recoveries. It can be seen from the figure that when the number of bars increased from 2 to 8, the recoveries of the target analytes increased. This result may be because the large volume extraction solvent can improve the extraction capacity of the target analytes. When the number of magnetized solvent-bars increased from 8 to 10, the recoveries decreased. The reason may be that in the limited space too many bars rub each other, which eventually causes loss of extraction solvent. Thus, eight magnetized solvent-bars were selected in the subsequent experiments.

Effect of the number of magnetized solvent-bars. Extraction solvent, chloroform; extraction time, 20 min; extraction temperature, 30 °C; stirring speed, 700 rpm; NaCl concentration, 10 %; pH of sample solution, 3.0
Effect of Extraction Time
The principle of HF-MSBLPME is based on the partitioning of the analyte between the aqueous sample and the organic solvent. It is an equilibrium process rather than an exhaustive extraction. It requires a period of time for equilibrium to be established. Nonetheless, too long an extraction time is unnecessary, as long as equilibrium can be established. On the other hand, if the extraction time is too long, solvent loss and formation of air bubbles may occur, which is not beneficial to the recoveries. Hence, the effect of extraction time on recoveries was investigated in the range of 5–25 min. The experimental results showed that the recoveries of the analytes increased significantly when the extraction time increased from 5 to 20 min, and after 20 min the recoveries appeared to be in a slow descent. Therefore, 20 min was chosen to be the extraction time.
Effect of Extraction Temperature
Extraction temperature is another significant factor. In this work, the effect of extraction temperature ranging from 25 to 45 °C was studied. The experimental results showed that the recoveries of the target analytes increased with increase of the extraction temperature, and the recoveries reached the highest level at 30 °C. When the extraction temperature was higher or lower than 30 °C, the extraction recoveries decreased. The reason may be that increasing temperature can result in the increase of the diffusion rate of analytes, and this is beneficial to the transfer of analytes into the acceptor solution. Nonetheless, too high a temperature may result in the evaporation of analytes from aqueous solution into the headspace, and it is not beneficial to the transfer of analytes into the extraction solvent. So, 30 °C was adopted for further experiments.
Effect of Stirring Speed
Stirring speed is also extremely important. Stirring the sample solution could enhance the mass transfer in the aqueous phase and reduce the extraction time to achieve the extraction equilibrium. Thus, the equilibrium of the analytes between the aqueous and organic phases could be achieved more rapidly by stirring the aqueous sample. Unfortunately, high stirring speed generated some problems, such as promotion of solvent evaporation and production of air bubbles on the surface of the hollow fiber. To attain optimal stirring speed, the effect of stirring speeds ranging from 400 to 800 rpm was studied. The results are shown in Fig. 4. It is seen from Fig. 4 that the recoveries increased with the stirring speed up to 700 rpm, and after 700 rpm the recoveries showed a slow descent. Thus, the stirring speed of 700 rpm was chosen for subsequent experiments.

Effect of stirring speed. Extraction solvent, chloroform; extraction time, 20 min; extraction temperature, 30 °C; number of magnetized solvent-bars, 8; NaCl concentration, 10 %; pH of sample solution, 3.0
Effect of NaCl Concentration
The effect of NaCl concentration on the recoveries was tested by adding NaCl solution (0–30 %, w/v) into the sample solution. The results showed that the recoveries increased with the increase of NaCl concentration from 0 % to 10 %. The reason may be that the addition of NaCl could give rise to the increase in the ionic strength of the solution and the decrease of the solubility of the analytes in the aqueous phase. The recoveries decreased with the increase of NaCl concentration from 10 % to 30 %. It is possible that the high NaCl concentration can result in the decrease of diffusion rate of the analyte into the organic phase. Based on the experimental results, 10 % NaCl was selected in the subsequent experiments.
Effect of pH of Sample Solution
In order to obtain efficient extraction of target analytes, which are weakly acidic, the sample solution should be adjusted to a suitable acidity to deionize analytes and maintain their neutrality to improve their distribution into the organic solvent. The pH value of a sample solution was adjusted with 0.1 mol/L HCl solution. The effect of pH in the range of 2.0–6.0 on recoveries was investigated. As shown in Fig. 5, the highest recoveries could be obtained at pH 3.0. Therefore, the pH value of 3.0 was chosen as the optimum value of pH for the extraction.

Effect of pH. Extraction solvent, chloroform; extraction time, 20 min; extraction temperature, 30 °C; stirring speed, 700 rpm; NaCl concentration, 10 %; number of magnetized solvent-bars, 8
Method Validation
The method was evaluated in terms of the linearities, limits of detection (LODs) and quantification (LOQs), and recoveries.
Analytical Performances
The working curves were made by plotting the peak areas measured versus the concentrations of analytes in the spiked samples under the optimized conditions. The procedure was carried out in triplicate for each concentration. The linear regression equations, linear ranges, and correlation coefficients are listed in Table 1. The LODs and LOQs indicated in Table 1 were determined at a signal-to-noise (S/N) ratio of 3 and 10, respectively. As shown in Table 1, the intercept of nicosulfuron is much higher than those of other analytes and the slope of nicosulfuron is lower than those of other analytes. The high intercept is mainly due to the low slope. The LOD of nicosulfuron was slightly higher than those of other analytes (Table 1), and most RSDs at a low concentration of nicosulfuron in spiked samples were higher than those of other analytes (Table 2).
Analysis of Samples
In order to validate the applicability of the newly established analytical method, four kinds of pear samples were analyzed. The sulfonylurea herbicides in these pears were not detectable. The practical applicability of the present method was evaluated by determining four sulfonylurea herbicides from fresh spiked samples. The analytical results are listed in Table 2. It can be seen that the recoveries range from 83.6 % to 100.1 % with RSDs ranging from 1.3 % to 6.8 %. The results show that the present method provides acceptable recoveries and precision for determination of sulfonylurea herbicides in pears. The HPLC chromatograms of both the blank and the spiked sample are shown in Fig. 6. The spiked concentration of each analyte is 250 ng/g. The pretreatment procedure for the samples are indicated in Sects. 2.3 and 2.4.

Chromatograms of the blank sample 1 (a) and the spiked sample 1 (b)
Comparison of the Present Method with Other Methods
To evaluate the present method, a comparison of the present method and other methods reported in literature was made. The results are shown in Table 3. The recoveries were acceptable. Compared with other methods, the present method was simpler and has some advantages in volume of the organic extraction solvent and the extraction time. Considering the advantages, HF-MSBLPME should be a satisfactory method.
Conclusions
In this paper, a novel HF-MSBLPME was successfully applied to the extraction of four sulfonylurea herbicides in pears. The magnetized solvent-bar was easy to make, and permitted active magnetic stirring, extraction, and pre-enrichment in a single device simultaneously. The magnetized solvent-bar was also easy to retrieve after extraction with a magnet, and the extract is conveniently collected. Compared with other methods, the present method has some advantages in the expenditure of the organic extraction solvent volume and the extraction time. The experimental results showed that HF-MSBLPME can be successfully applied to the extraction of sulfonylurea herbicides in fruits.
References
Fang GZ, Chen J, Wang JP, He JX, Wang S (2010) J Chromatogr A 1217:1567–1574
Carabias-Mart´ınez R, Rodr´ıguez-Gonzalo E, Herrero-Hernández E, Hernández-Méndez J (2004) Anal Chim Acta 517:71–79
She YX, Cao WQ, Shi XM, Lv XL, Liu JJ, Wang RY, Jin F, Wang J, Xiao H (2010) J Chromatogr B 878:2047–2053
Yan CM, Zhang BB, Liu WY, Feng F, Zhao YG, Du H (2011) J Chromatogr B 879:3484–3489
Yang MX, Zhang YY, Lin S, Yang XL, Fan ZJ, Yang LX, Dong XC (2013) Talanta 114:143–151
Zhou QX, Liu JF, Cai YQ, Liu GG, Jiang GB (2003) Microchem J 74:157–163
Peng Y, Xie Y, Luo J, Nie L, Chen Y, Chen L, Du SH, Zhang ZP (2010) Anal Chim Acta 674:190–200
Wu QH, Wang C, Liu ZM, Wu CX, Zeng X, Wen JL, Wan Z (2009) J Chromatogr A 1216:5504–5510
Fang GZ, Wang XN, Wang S (2010) Chromatographia 72:403–409
Chen ZL, Kookana RS, Naidu R (2000) Chromatographia 52:142–146
Asiabi H, Yamini Y, Moradi M (2013) J Supercritical Fluids 84:20–28
Howard AL, Taylor LT (1992) J Chromatogr Sci 30:374
Zhang P, Buia A, Rosea G, Allinson G (2014) J Chromatogr A 1325:56–64
Seccia S, Albrizio S, Fidente P, Montesano D (2011) J Chromatogr A 1218:1253
Tang KJ, Chen SW, Gu XH, Wang HJ, Dai J, Tang J (2008) Anal Chim Acta 614:112
Font N, Hernandez F, Hogendoorn EA, Baumann RA (1998) J Chromatogr A 798:179
Pedersen-Bjergaard S, Rasmussen KE (1999) Anal Chem 71:2650
Payan MR, Lopez MAB, Fernandez-Torres R, Navarro MV, Mochon MC (2011) J Chromatogr B 879:197–204
Tao Y, Liu JF, Hu XL, Li HC, Wang T, Jiang GB (2009) J Chromatogr A 1216:6259–6266
Ramos-Payán M, Bello MA, Fernández-Torres R, Pérez-Bernal JL, Callejón M (2009) Anal Chim Acta 653:184
Psillakis E, Kalogerakis N (2003) Trends Anal Chem 22:565
Rasmussen KE, Pedersen-Bjergaard S (2004) Trends Anal Chem 23:1
Pedersen-Bjergaard S, Rasmussen KE (2005) J Chromatogr B 817:3
Pedersen-Bjergaard S, Rasmussen KE (2008) J Chromatogr A 1184:132
Jiang XM, Lee HK (2004) Anal Chem 76:5591–5596
Baltussen E, Cramers CA, Sandra PJF (2002) Anal Bioanal Chem 373:3–22
Ridgway K, Lalljie SPD, Smith RM (2007) J Chromatogr A 1153:36–53
Xu X, Liang FH, Shi JY, Zhao X, Liu Z, Wu LJ, Song Y, Zhang HQ, Wang ZM (2013) Anal Chim Acta 790:39–46
Carabias-martinez R, Miranda-cruz E (2007) Electrophoresis 28:3606–3616
Ou XM (2006) Fine Chemical Intermediates 1:1–6
Haginaka J (2009) J Sep Sci 32:1548–1565
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shi, J., Li, X., Liu, C. et al. Determination of Sulfonylurea Herbicides in Pears Using Hollow Fiber-Protected Magnetized Solvent-Bar Liquid-Phase Microextraction HPLC. Chromatographia 77, 1283–1290 (2014). https://doi.org/10.1007/s10337-014-2740-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-014-2740-7