
Abstract
A method of hollow fiber–stir bar sorptive extraction (HF–SBSE) coupled with gas chromatography–mass spectrometry was proposed to explore the migration of eight organochlorine pesticide residues in environmental and biological matrices. HF–SBSE was a configuration for solid–phase microextraction purposes, combining the features of SBSE and hollow fiber membrane extraction technology. Variables affecting the extraction (salt addition, extraction temperature, and time) and desorption (microwave time, eluted solvent) have been evaluated. Under the optimal conditions, good linearity (r > 0.999) of all calibration curves was obtained in validation experiments. The recoveries in different sample matrices were in the range from 69.0 to 114.1% with RSD% less than 10.2%. The bio-accumulation of OCPs was tested in samples of soil, underground water, forage grass, and raw milk. It was indicated that HF-SBSE could be applied in simultaneous extraction of hazardous substances in different matrices without sample clean-up.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The contamination of the environment with pesticide residues is universal. Among the residues, the detection of the organochlorine pesticides (OCPs) was high (Tanabe et al. 1994; Wania and Mackay 1996). The determination of organochlorine pesticides in environmental samples and biological matrices has been a major subject for many years because of their potential risk for human health, persistence and tendency to bio-accumulate (Chiuchiolo et al. 2004; Nakata et al. 2005). Food chain was a major mechanism of OCP transfer from environment to biota (Tsygankov et al. 2015). It is necessary to carry out extensive monitoring programs for evaluating the levels of OCPs both in environmental and biological samples as indicators of possible migration pathways.
Studies involving the determination of OCPs in complex matrices often deal with samples with low analyte level and a large number of interfering substances. Traditionally, the determination of OCPs usually comprises three steps (Xue et al. 2008; Oh 2007): sample clean-up, extraction and chromatographic analysis, which was time-consuming and labor intensive. There were various types of pretreatment methods for pesticides, including solid–phase microextraction (Hwang and Lee 2000; De Carvalho et al. 2009a, b), matrix solid–phase dispersion microextraction (Zhao et al. 2006; De Carvalho et al. 2009a, b; Tang et al. 2006), and stir bar sorptive extraction (Ochiai et al. 2011; Rodil and Popp 2006). Use of hollow fiber membranes coated with functional polymers for the microextraction of organochlorine pesticide residues was also proposed by Lee’s group (Basheer et al. 2004; Basheer et al. 2007).
As one of the most widely used sample pretreatment techniques for liquid samples, solid–phase microextraction (SPME) has been increasingly and widely used to determine trace or ultra-micro-levels of both inorganic and organic analytes from different complex samples (Arthur and Pawliszyn 1990; Ouyang and Pawliszyn 2006; Ballesteros–Gőmez and Rubio 2011). However, further development of the technology in SPME area is still full of great challenges, and there is a tendency to combine different processes into one single hybrid processes (Tang et al. 2016; Li et al. 2015; Pabby and Sastre 2013). A combination of several configurations is a feasible way to develop a new extraction approach, which may synergistically derive advantages from existing individual methods, yet with innovative merits. Hollow fiber–stir bar sorptive extraction (HF–SBSE) is a kind of combination configuration of SBSE and hollow fiber membrane extraction.
The aim of the present study was to develop a reliable and robust alternative enrichment method based on a hollow fiber–stir bar sorptive extraction for the quantification of organochlorine pesticide residues in complex matrices. The main feature of HF–SBSE was the use of microporous HF acting as the carrier and filter, while a thin stainless steel wire and microspheres powder loaded in the lumen of HF respectively acting as the magnetic stirrer and the sorbents for the collection and extraction of OCP residues, thus affording extraction process like SBSE. ZrO2/SiO2 composite microspheres were fabricated by sol–gel method and served as the sorbent material to extract OCPs, and the porous hollow fiber membrane was used as filtration membrane to minimize the chromatographic interferences with the matrices. HF–SBSE method without sample clean-up has also been applied to determine the migration of OCPs from the environmental to biological matrices.
Experimental
Reagents and Materials
Tetraethyl orthosilicate (TEOS), zirconyl chloride octahydrate (ZrOCl2·8H2O), hydrogen peroxide (30%), all of analytical grades (purity > 99.9%), were procured from National Institute for Control of Pharmaceutical and Biological Products (Beijing, China). The mixed standards, benzene hexachloride (BHC, including α-BHC, β-BHC, γ-BHC, δ-BHC), 1,1-dichloro-2,2-bis(p-chloro-phenyl)ethylene(p,p′-DDE), 1,1-dichloro-2,2-bis(p-chloro-phenyl)ethane(p,p′-DDD), 1,1,1-trichloro-2-(o-chlorophenyl)-2-(p-chlorophenyl)ethane (o,p’-DDT), and 1,1,1-trichloro-2,2-bis (p-chlorophenyl)ethane(p,p′-DDT), were procured from Agro-Environmental Protection Institution (Tianjin, China). Methanol, ethanol, ethyl acetate, n-hexane, acetone, dimethylformamide (DMF), chloroform, hydrochloric acid, ammonia, sodium chloride, all of analytical grades, were procured from Tianjin No.3 Chemical Reagent Factory (Tianjin, China). The polyvinylidene fluoride (PVDF) hollow fiber membrane (900-μm i.d., 300-μm wall thickness, and 0.02-μm pore size, pH 2–11) was bought from Haike Membrana Co. Ltd. (Guangzhou, China). High-purity deionized water was used throughout the whole experiments. The soil, underground water, forage grass, and raw milk were sampled from the pasture land in Yuzhong area of Lanzhou, China.
Apparatus
GC–MS, thermo Scientific DSQ™ II (Thermo Fishier Scientific, Assembled in China) equipped with splitless injector, was employed for organochlorine pesticide residues. A Thermo TR-5 MS column, 30 m × 0.25 mm i.d., 0.25-μm film thicknesses (Thermo, USA) was applied to analyze the extracted analytes. Helium (99.999%) was used as the carrier gas and kept at a flow rate of 1.0 mL min−1. MS transfer line heater was set to 280 °C; ion source temperature was set to 250 °C; inlet temperature was set to 240 °C; EI was set to 70 eV. The GC–MS temperature program used was as follows: initial temperature 80 °C, first increased to 230 °C by 20 °C min−1 and held for 1 min, then raised to 270 °C by 20 °C min−1 and held for 2 min. Standards and samples were analyzed in selective ion monitor (SIM) mode. Samples were injected in a splitless mode. Major fragment ions for each organochlorine pesticide were listed in Table 1.
A JSM-5600 (JEOL, Tokyo, Japan) SEM system and a JFC-1600 Auto fine coater (JEOL, Tokyo, Japan) were used for the SEM experiments.
Preparation of Standards and Sample Solutions
For GC–MS analysis, the mixed stock solutions were diluted with n-hexane to give a concentration of 0.8~5 μg mL−1 for each analyte. The stock solution was diluted with n-hexane to obtain calibration standard solution, 0.0001, 0.001, 0.01, 0.1, 1, and 5 μg mL−1.
Samples collected from Yuzhong area of Lanzhou City, Gansu province, China, were chosen as the characteristic sample for test. It should be noted that HF–SBSE could only be directly applied in liquid samples. No pretreatment was required for liquid samples. But for dry samples, extraction with solvent should be carried out first. Dried solid samples (soil and forage grass) were extracted by chloroform (1:30, w/v) at 50 °C in an ultrasonic bath for three times. Not only did chloroform have a good solubility to OCPs, but also it can damage the cell structure, promoting the dissolution of OCPs in extracted solution. Soil and forage grass were homogenized in a stainless steel blender and sift through a 50-mesh sieve. Of the accurately weighed soil and forage grass powder sample, 5.0 g of the accurately weighed soil and forage grass powder samples were extracted with chloroform separately in an ultrasonic bath for 30 min. The extract solution was then filtered through a filter paper and transferred into a 250-mL volumetric flask, and preserved in refrigerator. Extracted solutions were applied as liquid samples to perform HF–SBSE.
Preparation of Hollow Fiber Stir Bar
Preparation of Core–Shell SiO2/ZrO2 Composite Microspheres
Monodisperse SiO2 microspheres were synthesized by a modified Stöber method (Büchel et al. 1998; Li et al. 2011). To prepare samples for SEM sample, the ZrO2/SiO2 composite microspheres were fixed on the stub by a double-sided sticky tape and then coated with Aurum by Auto fine coater for 5 min. The average diameters of the silica particles were about 1.5 μm, as shown in Fig. 1a. Core–shell SiO2/ZrO2 composite microspheres was fabricated according to the reference (Xu and Lee 2007). SEM of shell–core ZrO2/SiO2 composite microspheres was shown in Fig. 1b, c. A uniform surface coating of ZrO2 particles can be visualized and almost no aggregation can be observed. The average particles sizes of the ZrO2/SiO2 composite microspheres were approximately 1.6 μm, slightly bigger than the uncoated SiO2 microspheres before.

SEM of ZrO2/SiO2 composite microspheres. a Silica core microspheres. b, c Shell–core ZrO2/SiO2 composite microspheres)
Preparation of Hollow Fiber Stir Bar
PVDF hollow fiber was cut for 2 cm by scissors and ultrasonically cleaned in acetone to remove any possible impurities in the fiber. One end of HF was sealed with a lighter. A stainless steel wire of 1 cm length was inserted in the lumen of HF. 0.1 g ZrO2/SiO2 composite microspheres were dispersed in 1 mL absolute ethanol under sonication. Then, the dispersion was injected slowly with a syringe into the lumen of hollow fiber sealed at one end. After ethanol was volatilized, the other end of HF was sealed with the lighter. The preparation of hollow fiber stir bar was illustrated in Fig. 2.

Schematic illustration of preparation of the hollow fiber stir bar
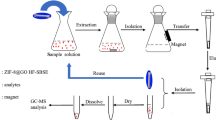
Extraction and Desorption Mode
Prior to use, hollow fiber stir bar was pre-rinsed with acetone and dried in order to remove impurities. All the liquid samples and extracted solutions were mixed with ethanol at the volume ratio of 2:1. The presence of ethanol can sharply decrease the surface tension of the matrices, which made the analytes facile to penetrate the pores of the PDVF hollow fiber membrane. The hollow fiber stir bar was first immersed in the mixed sample (2.0 mL) in a small glass vial under magnetic stirring for a certain time to extract OCPs. After extraction, stir bar was collected carefully by tweezers, placed in a centrifuge tube, and the analytes were eluted from stir bar by an appropriate organic solvent (1.00 mL) under microwave irradiation (700 W) for 2 min. Microwave irradiation contributed to the full and rapid desorption of OCPs into the eluted solvent. One-microliter eluted solution was directly injected into the GC–MS system for analysis.
Since the length (about 1.2 cm) of the hollow fiber stir bar used was short and only a small amount of composite microspheres were needed for filling-in, the stir bar can be considered disposable; little was gained from having to regenerate, recondition, and reuse the fiber. The low cost of the fiber was another advantage. Thus, the extraction device was considered for single use only and sample carryover effects were not an issue in this technique.
Results and Discussion
HF–SBSE was an equilibrium-based extraction procedure operating under the principle of organic analytes partitioning between the sample and the sorbents loaded inside of the fiber lumen. The ZrO2 nanoparticles deposited on the surface of SiO2 microspheres were expected to enhance the extraction efficiency. The analytical factors affecting efficiency such as ionic strength of the samples, extraction time and temperature, microwave desorption time, type of eluted solvent, and stirring rate were optimized.
Property of ZrO2 Nanoparticles
Silica was a widely used inorganic sorbent material. The reasons of depositing ZrO2 nanoparticles on the surface of nonporous SiO2 microspheres were as follows: (1) Zirconia was an amphoteric metal oxide, which exhibited both anion- and cation-exchange properties depending on the solution pH and the nature of the buffer (Rigney et al. 1990). A large number of strong Lewis acid sites on zirconia surface can interact with Lewis bases as R–SO3−, R–PO3−, R–COO− groups, etc. (Nawrocki et al. 1993). While OCP residues started contacting with ZrO2 nanoparticles, strong interaction was formed between inorganic adsorbents with trapped compounds; (2) the specific surface area and extraction sites of microspheres were increased by the ZrO2 nanoparticles depositing on it. Therefore, shell–core ZrO2/SiO2 composite microspheres had better extraction capacities than the core of silica. The ZrO2/SiO2 composite microspheres, as one kind of inorganic adsorbent materials prepared by sol–gel methods, were chemically stable in both acid and alkali matrices. It was also applied as the sorbent in chromatography column of HPLC (Li et al. 2011). Therefore, it would not be deformed or collapsed under high pressure. Therefore, the composite microspheres were stable under the extraction condition.
Optimization of Extraction Process
To avoid interfering with the matrix effect, the standard solution was used when optimizing the factors. HF–SBSE was an equilibrium extraction procedure. The amount of OCPs extracted depended on the partition coefficient, as well as the rate of mass transfer process at the interface of the aqueous phase (i.e., sample) and adsorbent phase (i.e., ZrO2/SiO2 composite microspheres). Extraction time and temperature were interrelated variables, and their effect on the HF–SBSE process should not be examined by simply changing one variable at a time (Stashenko and Martínez 2007). In our experiment, the extraction equilibrium was simultaneously established within the range of 10–60 min and 10–60 °C. The amount of p,p′-DDE (DDE was taken as an example for illustration, other OCPs were similar) generally increased with extraction time up to 30 min and with extraction temperature up to 40 °C (see Fig. 3a) and then no significant increase was observed thereafter. Thus, an optimum extraction time of 30 min and temperature of 40 °C were selected at the same time for subsequent experiments.

a Effects of extraction time and temperature on the extraction efficiency (p,p′-DDE was taken as an example for illustration). Conditions: 12 mm hollow fiber stir bar, sample volume 1.0 mL. b Effects of microwave desorption time on the desorption efficiency. Conditions: 12 mm hollow fiber stir bar, sample volume 1.0 mL, extraction time 30 min, extraction temperature 40 °C. c Effects of eluted solvents on the desorption efficiency. Conditions: 12 mm hollow fiber stir bar, sample volume 1.0 mL, extraction time 30 min, extraction temperature 40 °C. d GC–MS of different unspiked sample and standard solution (0.01 μg mL−1) analyzed by HF–SBSE method (1: α-BHC, 2: β-BHC, 3: γ-BHC, 4: δ-BHC, 5: p,p′-DDE, 6: p,p′-DDD, 7: o,p′-DDT, 8: p,p′-DDT)
For many analytes, aqueous solubility of analytes decreases as ionic strength of matrices increases; thus, theoretically, an enhancement of their extraction from the aqueous solution could be observed. The influence of salt on the extraction efficiency of pesticides using HF–SBSE was investigated by adding various amounts of NaCl (ranging from 5 to 30%, w/v). However, the addition of NaCl did not improve the extraction efficiency significantly. In other words, the solubility of OCPs was not sensitive to the concentration of salt. Therefore, no salt was added to improve the ionic strength of samples.
Optimization of Desorption Process
Eluted Solvents
Theoretically, OCP residues have hydrophobic functional group and tend to dissolve in the solvent with lower polarity. In the optimization of desorption, different solvents with a wide polarity range, such as n-hexane, acetone, chloroform, ethyl acetate, DMF, and methanol were studied as eluted solvents to desorb OCP residues from the stir bar under microwave assistance. Efficiencies of eluted solvents were compared in Fig. 3b. It was clear that chloroform achieved the highest peak areas for most OCPs, probably due to the principle of “like dissolves like.”
Microwave Desorption Time
Inorganic adsorbents interacted strongly with trapped compounds and required relatively high desorption temperature. Usually, water bath and sonication were used to assist desorption of OCPs from inorganic oxide under high temperature. The advantages of microwave were heating rate acceleration, milder conditions, and lower energy usage. Since OCP residues were stable to temperature, microwave was used for rapid heating to destroy the interaction between inorganic adsorbents and the analytes. The microwave time was set from 30 to 180 s at the interval of 30 s. It turned out that the peak areas of OCPs in Fig. 3c increased with the microwave time up to 120 s and then no significant increase was observed thereafter. Two minutes appeared to reach the desorption equilibrium for all analytes.
Besides, the effect of stirring rate on the extraction efficiency of target analytes was also studied within 500–900 rpm. The experimental results indicated that the extraction efficiency of OCPs increased with the increase of stirring rate from 500 to 800 rpm and then adsorption equilibrium was attained with no further increase of stirring rate to 900 rpm. Finally, stirring rate of 800 rpm was selected in the work.
Method Validations
The linearity of OCP calibration plot was investigated with standard solutions by HF–SBSE method over a concentration range of 0.0001~5 μg mL−1. The correlation coefficient (r) for each analyte was greater than 0.999, indicating good linearity. The limits of detection (LODs) and limits of quantification (LOQs) (seen in Table 1) were calculated at S/N = 3 and S/N = 10 from the chromatograms of standards at low concentration levels.
The precision of the chromatographic peak areas was studied by five replicate experiments using standard solution containing 0.08~0.5 μg mL−1 of analytes. The relative standard deviation ranges from 1.9 to 5.0%, n = 5. For investigation of the preparation repeatability, five hollow fiber stir bars prepared in one batch and five stir bars prepared in five batches were employed for the extraction of the target analytes from the same standard solutions above. The calibration curves and relative standard deviations (RSDs) were listed in Table 1. The experimental data demonstrated that a satisfactory repeatability was achieved for preparation of stir bars in one batch (RSDs, 4.3–8.2%, n = 5) and batch to batch (RSDs, 3.2–8.5%, n = 5).
Besides, the LODs and LOQs for each matrices (soil, underground water, forage grass, and milk) were also investigated by HF–SBSE method. Liquid sample and extracted solution of dried sample (both 1.00 mL) were fortified with 0.0002~0.001 μg mL−1 standard solution of OCPs at the volume ratio of 1:1, and then to perform HF–SBSE procedure at above optimum conditions. LODs and LOQs of each matrix were also calculated at S/N = 3 and S/N = 10 from the chromatograms of spiked samples. The results were shown in Table 2.
The accuracy was confirmed by recovery test with standard addition method. Liquid samples and extracted solutions of dried samples (both 1.00 mL) were mixed with three levels of standard solution ranged from 0.0001 to 0.05 μg mL−1 for analytes in the volume ratio of 1:1, and then carried out with HF–SBSE method. Table 3 listed the absolute recoveries of the extracted analytes. Recoveries of four samples were satisfactory (between 69.0 and 114.1%, n = 5) for target analytes at the spiking levels. It should be mentioned that the recoveries of δ-BHC in all kinds of samples were a bit low than expected, probably because δ-BHC was strongly bonded to some components in the matrix or ZrO2/SiO2 microspheres, causing the decrease in recovery. The results mainly demonstrated that HF–SBSE was reliable for the analysis of OCP residues in complex samples.
Application to the Real Samples
Evaluation of the role that pollutants played in ecosystems and assessment of their influence on wildlife were possible only if the pathways by which pollutants get into animal organisms were taken into account. Therefore, the migration of OCP residues from environment to biota was tested in samples of soil, underground water, forage grass, and raw milk. The aim was to figure out the distribution curves of OCPs and try to propose the bio-accumulation process for OCP residues from environment to biota. Several kinds of target OCP residues were detected; the data in Table 4 showed pesticide residue levels in these detected samples. Figure 3d was GC–MS–SIM of unspiked sample and standard solution analyzed by HF–SBSE method. Thanks to the micropores provided by the membrane of HF stir bar, a relatively clean baseline can be obtained in the determination of all the samples at the quantitative concentration level. A certain amount of p,p′-DDE was quantified in underground water. Since OCPs were lipophilic compounds, they tended to be accumulated in lipophilic organ and samples. Therefore, p,p′-DDD was relatively high in raw milk, while it could only be detected in soil and forage grass samples. The results partly explained the bio-accumulation of some OCPs existed in milk samples.
And these concentrations of OCPs in real samples were also confirmed by the methods regulated in Chinese National Standard (issued by General Administration of Quality Supervision Inspection and Quarantine of the People’s Republic of China, GB/T 23200.86–2016: National food safety standards–Determination of multiple residue of organochlorine pesticides in milk and dairy products–gas chromatography-mass spectrometry; GB/T5009.146–2008: Determination of organochlorine and pyrethroid pesticide multi-residues in vegetable foods; DB22/T 2084–2014: Determination of organochlorine pesticides residues in soil–gas chromatography-mass spectrometry; HJ 699–2014: Water quality–Determination of organochlorine pesticides and chlorobenzenes–Gas chromatography mass spectrometry). Most concentrations of OCPs in real samples were very close by HF–SBSE and methods in Chinese National Standard. Except for several specific OCP concentrations in these samples, i.e., p,p′-DDE in milk, p,p′-DDD in soil, and α-BHC in underground water, which were formerly beyond the LOQs of HF–SBSE, were detected by methods in Chinese National Standard. The results in Table 4 basically indicated that HF–SBSE combined with GC–MS was a very promising tool for the rapid detection of low levels of OCPs in complex matrices.
Conclusions
The outstanding advantage of HF–SBSE in complex matrices was the prevention of direct contact of the sorbents with macromolecular impurities in the sample, and therefore preventing the contamination of the sorbents with interfering biological substances in the matrices and avoiding strong matrix effects. The high porosity of the HFs even allowed the coating of a large amount of polymers contributing to a high loading capacity and selectively for different types of analytes. Moreover, the sorbents in the stir bar can also be changed to self-made or commercialized sorbents according to affinities of the analytes with different polarity and structures. The conditions for extraction and elution were relatively mild, especially suitable to temperature-sensitive analytes. It is clear that further progress in analytical methods and developments in advanced materials could extend the applicability of this method.
This was the first application of HF–SBSE for the analysis of organochlorine pesticide residues in environmental and biological matrices. The results clearly showed that the ZrO2/SiO2 composite microspheres can be adopted as an effective sorbent for the simple and rapid enrichment of OCPs from complex samples and it may have a great application potential for the preconcentration of other analytes with different polarity. The results also revealed that OCP residues tended to be accumulated in lipophilic samples. Bio-magnification of harmful substances, i.e., their accumulation to high concentrations, in organisms of higher trophic levels is a manifestation of this process. The environment remained a natural reservoir receiving pollutants from various sources, and thus it required a regular monitoring of the state of the animal living in it. The strict regulation and monitoring of OCPs would be helpful in controlling the concentration of OCPs in the environment.
References
Arthur CL, Pawliszyn J (1990) Solid phase microextraction with thermal desorption using fused silica optical fibers. Anal Chem 62:2145–2148
Ballesteros–Gőmez A, Rubio S (2011) Recent advances in environmental analysis. Anal Chem 83:4579–4613
Basheer C, Suresh V, Renu R, Lee HK (2004) Development and application of polymer-coated hollow fiber membrane microextraction to the determination of organochlorine pesticides in water. J Chromatogr A 1033:213–220
Basheer C, Vetrichelvan M, Valiyaveettil S, Lee HK (2007) On-site polymer-coated hollow fiber membrane microextraction and gas chromatography–massspectrometry of polychlorinated biphenyls and polybrominated diphenylethers. J Chromatogr A 1139:157–164
Büchel G, Unger KK, Matsumoto A, Tsutsumi K (1998) A novel pathway for synthesis of submicrometer-size solid core/mesoporous Shell silica spheres. Adv Mater 10:1036–1038
Chiuchiolo AL, Dickhut RM, Cochran MA, Ducklow HW (2004) Persistent organic pollutants at the base of the Antarctic marine food web. Environ Sci Technol 38:3551–3557
De Carvalho PHV, Barreto AS, Rodrigues MO, Prata VD (2009a) Two-dimensional coordination polymer matrix for solid-phase extraction of pesticide residues from plant Cordia salicifolia. J Sep Sci 32:2132–2138
De Carvalho PHV, Prata VD, Alves PB, Navickiene S (2009b) Determination of six pesticides in the medicinal herb Cordia salicifolia by matrix solid-phase dispersion and gas chromatography/mass spectrometry. J AOAC Inter 92:1184–1189
Hwang BH, Lee MR (2000) Solid-phase microextraction for organochlorine pesticide residues analysis in Chinese herbal formulations. J Chromatogr A 898:245–256
Li J, Zhang H–F, Shi Y–P (2011) In-situ synthesis and normal chromatographic properties of nonporous SiO2/ZrO2 core–shell composite microspheres. Anal Sci 27:447–450
Li J, Wang YB, Li KY, Cao YQ, Wu S, Wu L (2015) Advances in different configurations of solid-phase microextraction and their applications in food and environmental analysis. Trend Anal Chem 72:141–152
Nakata H, Hirakawa Y, Kawazoe M, Nakabo T, Arizono K, Abe S–I, Kitano T, Shimada H, Watanabe I, Li W, Ding X (2005) Concentrations and compositions of organochlorine contaminants in sediments, soils, crustaceans, fishes and birds collected from Lake Tai, Hangzhou Bay and Shanghai city region, China. Environ Pollut 133:415–429
Nawrocki J, Rigney MP, McCormick A, Carr PW (1993) Chemistry of zirconia and its use in chromatography. J Chromatogr A 657:229–282
Ochiai N, Ieda T, Sasamoto K, Takazawa Y, Hashimoto S, Fushimi A, Tanabe K (2011) Stir bar sorptive extraction and comprehensive two-dimensional gas chromatography coupled to high-resolution time-of-flight mass spectrometry for ultra-trace analysis of organochlorine pesticides in river water. J Chromatogr A 1218:6851–6860
Oh CH (2007) Monitoring of residual pesticides in herbal drug materials of Korea and China. Bull Environ Contam Tox 78:314–318
Ouyang G, Pawliszyn J (2006) Configurations and calibration methods for passive sampling techniques. Trend Anal Chem 25:692–703
Pabby AK, Sastre AM (2013) State-of-the-art review on hollow fibre contactor technology and membrane-based extraction processes. J Membr Sci 430:263–303
Rigney MP, Funkenbusch EF, Carr PW (1990) Physical and chemical characterization of microporous zirconia. J Chromatogr A 499:291–304
Rodil R, Popp P (2006) Development of pressurized subcritical water extraction combined with stir bar sorptive extraction for the analysis of organochlorine pesticides and chlorobenzenes in soils. J Chromatogr A 1124:82–90
Stashenko EE, Martínez JR (2007) Sampling volatile compounds from natural products with headspace/solid-phase micro-extraction. J Biochem Biophys Meth 70:235–242
Tanabe S, Iwata H, Tatsukawa R (1994) Global contamination by persistent organochlorines and their ecotoxicological impact on marine mammals. Sci Total Environ 154:163–177
Tang F, Yue YD, Hua RM, Cao HQ (2006) Matrix solid–phase dispersion microextraction and determination of pesticide residues in medicinal herbs by gas chromatography with a nitrogen-phosphorus detector. J AOAC Inter 89:498–502
Tang S, Zhang H, Lee HK (2016) Advances in sample extraction. Anal Chem 88:228–249
Tsygankov VY, Boyarova MD, Lukyanova ON (2015) Bioaccumulation of persistent organochlorine pesticides (OCPs) by gray whale and Pacific walrus from the western part of the Bering Sea. Mar Pollut Bull 99:235–239
Wania F, Mackay D (1996) Peer reviewed: tracking the distribution of persistent organic pollutants. Environ Sci Technol 30:390A–396A
Xu L, Lee HK (2007) Zirconia hollow fiber: preparation, characterization, and microextraction application. Anal Chem 79:5241–5248
Xue J, Hao L, Peng F (2008) Residues of 18 organochlorine pesticides in 30 traditional Chinese medicines. Chemosphere 71:1051–1055
Zhao CJ, Hao GM, Li HX, Luo X, Chen YJ (2006) Decontamination of organochlorine pesticides in Radix Codonopsis by supercritical fluid extractions and determination by gas chromatography. Biomed Chromatogr 20:857–863
Acknowledgements
Financial supports from the National Nature Science Foundation of China (No. 21467027 No. 31260500, and No. 51563022) are gratefully acknowledged.
Funding
This study was funded by the National Nature Science Foundation of China (grant number 21467027).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Hong Li declares that he has no conflict of interest. Wen-Juan Zhang declares that she has no conflict of interest. Yan-Bin Wang declares that he has no conflict of interest. Qiong Su declares that she has no conflict of interest. Lan Wu declares that she has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Additional information
Chinese National Standard, General Administration of Quality Supervision Inspection and Quarantine of the People’s Republic of China, GB/T 23200.86–2016: National food safety standards–Determination of multiple residue of organochlorine pesticides in milk and dairy products–gas chromatography-mass spectrometry; GB/T5009.146–2008: Determination of organochlorine and pyrethroid pesticide multi-residues in vegetable foods; DB22/T 2084–2014: Determination of organochlorine pesticides residues in soil–gas chromatography-mass spectrometry; HJ 699–2014:Water quality– Determination of organochlorine pesticides and chlorobenzenes–Gas chromatography mass spectrometry
Rights and permissions
About this article

Cite this article
Li, J., Li, H., Zhang, WJ. et al. Hollow Fiber–Stir Bar Sorptive Extraction and Gas Chromatography–Mass Spectrometry for Determination of Organochlorine Pesticide Residues in Environmental and Food Matrices. Food Anal. Methods 11, 883–891 (2018). https://doi.org/10.1007/s12161-017-1053-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-017-1053-5