
Abstract
The aim of the present study was to develop a fast, sensitive and reliable method for rapid screening of cephalosporin injectable dosage forms namely ceftazidime and ceftizoxime to the detection of counterfeit and substandard drugs that might be illegally commercialized. Ceftazidime, ceftizoxime and cefixime (IS) were separated in a X-Terra RP-18 column (250 × 4.60 mm ID × 5 μ) and DAD detector set at 290 and 260 nm. The mobile phase consisted of a mixture of methanol:water 20:80 (v/v) at a flow rate of 1.0 mL min−1. Additionally, in order to find the optimum pH value of separation the pK a values of studied compounds were determined by using two different methodologies. Aqueous pK a values of studied compounds have been determined by UV-spectrophotometry and liquid chromatography were used for the determination and direct characterization of the dissociation constants by using the dependence of the capacity factor on the pH of the mobile phase in 20% (v/v) methanol–water binary mixture in which separation was performed. The pH of the mobile phase was adjusted with 25 mM H3PO4 to 3.2. The method was shown to be linear, sensible, accurate, and reproducible over the range of analysis and it can be used to pharmaceutical formulations containing a single active ingredient within a short analysis time.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cephalosporins are among the safest and the most currently administered parenteral agents available to the clinician, and have therefore become the most widely prescribed of all antibiotics [1]. Ceftazidime (CFT) and ceftizoxime (CFZ) are parenteral, third generation cephalosporin antibiotics that are administered by intravenous or intramuscular injection. CFT is effective against a wide variety of gram-negative organisms, including Pseudomonas aeruginosa. Although CFT is active in vitro against gram-positive organisms, clinical use of CFT for Gram (+) infections is rare. CFT is used for the treatment of meningitis, lower respiratory infections, febrile neutropenic events, urinary tract infections, pelvic inflammatory disease, and skin and skin structure infections.
Ceftizoxime is active against aerobic gram-positive bacteria, including staphylococci, aerobic Gram (−) bacilli and anaerobic bacteria. This broad-spectrum compound has a synmethoxyimino chain at the 7-position that confers, 3-lactamase stability [2]. Its other properties include poor induction of β-lactamase and a long serum half-life [3, 4]. Both drug active compounds cross the placenta and is distributed into breast milk.
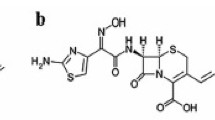
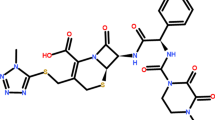
All of these semi-synthetic antibiotics derive from the 7-aminocephalosporanic acid composed of a β-lactam ring fused with a dihydrothiazine ring (Fig. 1), but differ in the nature of the substituents attached at the 3- and/or 7-positions of the cephem ring. These substitutions affect either the pharmacokinetic properties (3-position) or the antibacterial spectrum (7-position) of the cephalosporins [5]. Compared with the previous first- and second-generation agents, the third-generation cephapalosporins are characterized by a broader antibacterial spectrum, which is related to their enhanced stability to β-lactamases.

Chemical structure of studied compounds
Water solubility, partition coefficient, and dissociation constant are usually measured for characterizing physicochemical properties of drugs. Especially, for understanding and quantifying chemical phenomenon such as reaction rates, biological activity, biological uptake, biological transport, and environmental fate dissociation constants (i.e. pK a values) are a key parameter [6]. It has become of great importance because the passage of many drugs into cell and across other membranes is a function of the internal environment, of the physicochemical properties and of the pK a of the drugs [7]. A satisfactory knowledge of the acid–base behavior of substances in water is therefore essential. Moreover, the pH of the mobile phase is a major factor influencing the chromatographic behavior of ionogenic compounds because they contain functional groups such as carboxylic and aminothiazole center. Their retention depends on the percentage of ionized and non-ionized species of each compound. Although methanol (MeOH)–water mobile phases have been used in RP-LC separation procedures, the pK a values of CFT and CFZ have not yet been determined and reported in MeOH–water binary mixtures. Additionally, pK a values in hydroorganic media is therefore necessary to predict the influence of pH on selectivity and on retention in LC and also to optimize analytical procedures for the separation of ionizable compounds by different techniques [8, 9].
Several experimental approaches have been employed for the determination of the dissociation constants, including potentiometry, UV spectrophotometry, HPLC, phase solubility and recently capillary zone electrophoresis techniques [7, 10–14]. UV-spectrophotometric method hinges on neutral and ionic species having different spectra. When this criteria is met, excellent precision is obtained [15, 16]. The pK a determination with this method is also an attractive alternative, for concentrations over 10−6 mol L−1. Moreover, the true dissociation constants can be measured directly without inter-ionic effects having to be considered and the optical properties of the ions are virtually constant up to relatively high concentrations. However, in this technique, the studied compound should be pure and at least one of the species (acid or base) must contain an absorbing group.
Liquid chromatography is also used as a powerful technique for determination of dissociation constants due to requires only small quantity of compounds, studied samples do not need to be pure and poor water solubility is not a serious drawback. This method does not requires measuring solute or titrant concentrations, just only retention times. Also, calculation is straightforward and independent of solute purity. However, standard deviations of pKa values are higher than obtained in potentiometric and spectrophotometric methods.
HPLC has been used frequently in all fields of β-lactam research, not only as an assay method but also as a tool for purification of the antibiotics. Due to the insolubility of these compounds in organic solvents, normal phase LC was sparingly used. Most methods employ reversed-phase LC and chemically bonded packing materials. Literature survey reveals that the CFZ and CFT are not officially reported in any pharmacopoeia. While validated assays have been reported for each drug individually, there is no reported method that permits simultaneous quantification of CFZ and CFT in pure forms and pharmaceutical formulations [17–21].
This paper is focused mainly on the simple, rapid, accurate, precise and fully validated RP-LC method for the determination of CFT and CFZ in their parenteral dosage forms. Additionally, in order to find the optimum pH value of separation the pK a values of studied compounds were determined by using two different methodologies (UV spectrophotometry and liquid chromatography). The pK a values of studied compounds are either not known accurately or not available at all. Only a limited number of works related to pK a values of some of other cephaloporins are found in the literature [22–25]. Aqueous pK a values of studied drugs have been determined by spectrophotometric method and LC method was used for the determination of ionization constants in 20% (v/v) MeOH–water binary mixture in which separation was performed.
Experimental
Chemicals and Reagents
All chemicals were used without further purification. Cephalosporins used in this study were kindly supplied as follows: ceftizoxime (Zentiva Pharm. Ind. Istanbul), ceftazidime (Glaxosmithkline Pharm. Ind. İstanbul) and cefixime (IS) (Eczacıbaşı Pharm. Ind. İstanbul) (Fig. 1). MeOH was of HPLC grade from Merck (Darmstadt, Germany), ortho-phosphoric acid (min. 85%) were from Riedel (Riedel-de Haen, Germany). Sodium hydroxide, potassium hydroxide (Titrisol), hydrochloric acid (Titrisol), potassium hydrogen phthalate (dried at 110 °C before use) and potassium chloride (ionic strength adjuster; 0.1 mol L−1) were all supplied from Merck (Darmstadt, Germany). All stock solutions of hydrochloride acid, potassium chloride and potassium hydrogen phthalate were prepared by deionized water. Water used for the preparation of all aqueous solutions, with conductivity 18.2 μS cm−1 was obtained using a Zeneer Power I water system (Human Corp. Korea).
Apparatus
Potentiometric measurements were performed with Mettler-Toledo MA 235 pH/ion (resolution ±0.1 mV) analyzer system. All titrations were carried out under N2 and at 25.0 ± 0.1 °C, which was maintained by circulating water from a constant-temperature thermostat (Heto CBN 8-30 and temperature control unit Heto HMT 200) through the double-wall Pyrex titration cell of 80-ml capacity.
The UV–Vis absorbance spectra were recorded at each pH using Perkin Elmer LAMBDA 25 spectrophotometer, equipped with 1 cm path length cell, controlled by personal computer. A peristaltic pump equipped with the spectrophotometer was used to circulate the solution from the titration vessel to the spectrophotometer cell, and vice versa, through Teflon or Tygon tubes in a closed loop circuit with continuous flow.
The HPLC analysis was carried out on a Shimadzu HPLC system with a pump (LC-20 AD), a DAD detector system (SPD-M 20A) and column oven (CTO 20 AC). This equipment has a degasser system (DGU 20 A). A X Terra RP-18 column (250 × 4.60 mm ID × 5 μ) was used at 30 °C. Mettler Toledo MA 235 pH/ion analyzer with combined glass electrode was used for pH measurements of mobile phases. Potassium hydrogen phthalate was used as a reference value standard for the standardization of this apparatus in MeOH–water binary mixtures in accordance with IUPAC rules [26, 27].
Spectrophotometric Procedure
Before the spectrophotometric titration, carbonate-free potassium hydroxide solutions were prepared under a nitrogen atmosphere. The ionic strength of each KOH solution was adjusted to 0.10 mol L−1 by the addition of KCl. The alkali titre and absence of carbonate were periodically checked by pH-metry, using the appropriate Gran function against primary standard oven-dried potassium hydrogen phthalate [28].
The pK a values of the cephalosporins were determined by means of the data obtained from spectrophotometric titrations in water at 25.0 ± 0.1 °C and in 0.1 mol L−1 ionic strength (KCl). The spectrophotometric multiple-wavelength pH-titration was carried out as follows: in a first step, the standard emf values, E°, of the potentiometric cell were evaluated from titrations of a measured amount of an acidic solution, at the same conditions of temperature and ionic strength to be used in later experiments using KOH solutions in the same solvent and ionic strength as the titrant, and checking the calibration parameters from the Gran plots [28, 29]. The standard emf of the cell, E°, is the average of at least 15 standardizations.
In a second step, a solution of fully protonated drugs (32.0 mL containing 1.10−5 mol L−1 of drug) by HCl at the required conditions of temperature and ionic strength were titrated using KOH solutions in the same solvent and ionic strength in the pH range 1.2–12.0. After each addition, the potential was allowed to stabilise, its value used, in combination with E° calculated in calibration step, to calculate the pH of the solution. In the UV–Vis spectrophotometric titrations, the test solution was pumped to a spectrophotometric flow-cell by means of a peristaltic pump. After each addition of titrant, and after waiting for the emf reading to be stable, a spectrum, UV–Vis spectra were recorded with 1 nm resolution at 200–500 nm intervals in order to obtain different spectra around the maximum λ for each drug.
Chromatographic Procedure
Throughout this study, the mobile phases assayed were MeOH–water at 20, 25, and %30 (v/v), containing 25 mM o-phosphoric acid. The pH of the mobile phase was adjusted between 2.0 and 5.0 by the addition of sodium hydroxide. The flow rate was maintained at 1 mL min−1. For each drugs the retention time values, t R, were determined from three separate injections for each mobile phase composition and pH considered. Capacity factors were calculated as k = (t R − t 0)/t 0. The dead time (t 0) was measured by injecting uracil solution (0.01% w/v, in water), which was established for each mobile phase composition and pH studied. The chromatographic conditions were as follows: volume injected 20 μL, column temperature 30 °C, detection ceftizoxime and cefixime (IS) at 290 nm, and ceftazidime at 260 nm.
Preparation of Standard Solutions
Stock solution (100 μg mL−1) of cephalosporins was prepared in the mobile phase. All solutions were protected from light and were used within 24 h to avoid decomposition. The concentration of CFT and CFZ were varied in the range 1–16 and 1–20 μg mL−1, respectively, and the concentration of cefixime (IS) was maintained at a constant level of 4.0 μg mL−1. The calibration curves for LC analysis were constructed by plotting the ratio of the peak area of the drug to that of internal standard against the drug concentration.
Analysis of Parenteral Dosage Form
Cephalosporins based commercial parenteral dosage forms (flacons) were obtained from local pharmacy. Ceftazidime and ceftizoxime were provided by GlaxoSmithKline (Ceftazidime pentahydrate; Fortum® 1 g enjectable flacon) and Eczacıbasi Pharm. Comp. (Cefizox® 1 g enjectable flacon). An appropriate portion of these powders were weighted and stock solutions were prepared in mobile phase at a concentration of 100 μg mL−1 and the content of the flask was sonicated for 15 min. This solution was filtered and the filtrate was collected in a clean flask. Appropriate solutions were prepared by taking suitable aliquots of clear filtrate and adding the appropriate IS solution. The working standard solutions at the concentrations in the calibration range were prepared immediately before analysis by appropriate dilution of the stock solutions with the mobile phase. The amounts of ceftazidime and ceftizoxime were calculated from the corresponding regression equations. Standard stock solutions were stored at +4 °C for a week at most and their stability was checked over this period by daily preparing and injecting a diluted solution of each analyte.
Recovery Studies from Tablets and Laboratory-Made Mixtures
To verify the accuracy of the method, recovery experiments were performed by adding a known amount of pure drug to the preanalyzed flacons. Known amounts of the pure drug and at a constant level of IS were added to ceftazidime and ceftizoxime dosage forms and the mixtures were analyzed. The percent recovery was calculated by comparing the concentration obtained from spiked samples with the actual added concentration. Thus, the effect of common excipients in pharmaceutical dosage forms on chromatograms (e.g., tailing, broadening) was investigated. Recovery experiments also showed the reliability and suitability of the method. In order to demonstrate the applicability of the method, the recovery tests were also carried out by analyzing synthetic mixtures of the CFT and CFZ. After five repeated experiments, the recoveries from these synthetic mixtures were calculated for each compound.
Results and Discussion
In the present work, pK a values of CFT and CFZ were determined by two different methods and different solvents. The absorption spectra of CFT and CFZ in water at various pH values at 200–500 nm intervals were recorded and sample spectra of studied compounds are shown in Fig. 2. The spectrophotometric data are processed using the STAR program in order to obtain the pK a values for substances using iterative procedure [30].

Plot of experimental absorbance values of ceftazidime (a) and ceftizoxime (b) as a function of pH in water
For liquid chromatography, pK a values were determined from k/pH data pairs by means of the non-linear regression program NLREG [31]. This is a general purpose program, where the function to be minimized and the parameters to be estimated can be defined by means of the built-in program editor. Spectrophotometric values were recorded at 25.0 ± 0.1 °C, while RP-LC values are in 30.0 ± 0.1 °C. The examples of dependences of the capacity factors on the pH value in the mobile phase (20% v/v MeOH) are given in Fig. 3. The correlation between the experimental capacity factors of the compounds studied over the whole experimental pH range was good, as shown in Fig. 3.

Plots of the capacity factors versus the pH of the mobile phase for 20% (v/v) MeOH: a ceftazidime, b ceftizoxime
Ceftazidime contain a strong acidic carboxylic group directly attached on the cephem nucleus (about pK 1.9), a weak aminothiazole center in the side chain (pK a 4.1). CFT bears a second carboxylic group in the side chain, which further contributes to the complicated dissociation equilibria (about pK a 2.7) and a permanently charged quaternary nitrogen atom at position 3 of the cephem nucleus [32]. Over a broad pH range, they exist as zwitterions, formed between the carboxylate anion and the positively charged quaternary nitrogen atom. To a lesser extent, zwitterionic species formation is possible between the carboxylate anion and the protonated aminothiazole moiety in a limited pH range. As can be deduced from Fig. 3a, amonothiazole and carboxylic groups can be determined by chromatographic method. CFZ has an amino-thiazole moiety on its side chain at the C-7 position (pK a 2.95) [33]. Due to the limiting pH values of the stationary phase, only one pK a value can be determined for CFZ corresponds to the amino-thiazole moiety (Fig. 3b). The dissociation constant values determined for involved equilibria for the CFT and CFZ studied in methanol–water mixtures [20% (v/v)] are collected in Table 1, together with respective standard deviations.
Various mobile phase systems were prepared and used to provide an appropriate RP-LC separation (20, 25, and 30% v/v; MeOH–water mixtures). The experimental region was selected in such a way that the capacity factors of the studied compounds would stay within the limits 1 < k < 10. X Terra C-18 column has been selected and used successfully as a stationary phase for the simultaneous determination and well separation of CFT, CFZ, and IS. This stationary phase has an extended pH stability, to be thermally more stable, and to be more efficient than classical silica-based packing.
In order to find the optimum chromatographic conditions for RP-LC determination of CFT, CFZ, and IS, the influence of pH on the mobile phase and column temperature were examined. The optimum pH values for separation could be within the range of pK a ±1.5. Thus, pH 3.2 was selected as optimum pH value with best peak asymmetry and retention values. pH of the mobile phase has always been adjusted with 25 mM ortho-phosphoric acid. The column temperature was set at between 25 and 30 °C. The use of a higher temperature has been shown to reduce the tailing of the studied compounds. 30 °C was selected because shorter analysis time and improved peak shapes. Mobile phases containing the organic modifier methanol have been shown to improve peak performance. Finally, the mobile phase MeOH:water 20:80 (v/v) with 25 mM H3PO4 (at pH 3.2) at a flow rate of 1.0 mL min−1 was chosen as the most suitable carrier for RP-LC analysis. The proposed RP-LC method provides a simple procedure to simultaneous analysis of CFT and CFZ in drug formulations by DAD detection.
System suitability parameters for the proposed method were evaluated. The parameters tested for system suitability included retention time, tailing factor, capacity factor, resolution, theoretical plates, selectivity factor and RSD% of retention time. The results from system suitability tests are presented in Table 2 for the studied compounds. As can be deduced from Table 2, the presented chromatographic conditions ensure adequate retention of all compounds. Also the proposed method has enabled good resolution of analytes, since values of resolution factors of adjacent peaks were greater than 2.0. The results obtained from system suitability tests are in agreement with the USP requirements.
The calibration curves and equations for CFT and CFZ were calculated by plotting the peak area ratios of drugs to IS versus concentration of the compounds in the range 1.0–16.0 μg mL−1 and 1.0–20.0 μg mL−1, respectively (Table 3). These results showed highly reproducible calibration curves with correlation coefficients of >0.999. The low values of SE of slope and intercept and greater than 0.999 correlation coefficient for all compounds established the precision of the proposed methods. The LOD and LOQ were calculated from the following equations using the standard deviation of response (s) and the slope (m) of the corresponding calibration curve [34, 35].
The developed method was validated according to the standard procedures and the obtained results are given in Tables 3 and 4. Accuracy, precision, and reproducibility of the proposed method were assessed by performing replicate analysis of the standard solutions in mobile phase. Within calibration curves, three different concentrations were prepared for both compounds and assayed with related calibration curves to determine within-day and between-day variability. The within-day and between-day precision, accuracy, and reproducibility were determined as the RSD% and mean value. Precision, accuracy, and reproducibility results demonstrate good precision, accuracy, and reproducibility.
In order to demonstrate the validity and applicability of the proposed methods, recovery studies were performed by analyzing synthetic mixtures of CFT, CFZ, and IS which reproduced different composition ratios (Table 5).
A typical chromatogram is shown in Fig. 4, under the described experimental conditions, the peaks corresponding to the compound studied have symmetrical single peaks, forming well shaped, well resolved and perfectly separated from the solvent front and each other. Additionally chromatographic run was done within 12 min.

Chromatogram obtained from a standard mixture of studied compounds (each drug 8 μg mL−1). The chromatograms are monitored at 260 nm (A) and 290 nm (B). Ceftazidime (1), ceftizoxime (2), cefixime (3)
Precision and reproducibility of the method were performed by doing replicate analysis of standard solutions in the mobile phase. Repeatability and reproducibility were characterized by mean recovery and RSD% and the obtained results are given in Table 5. As can be deduced from Tables 4 and 5, there was no significant difference for the assay, as tested by within-day and between days.
When working on standard solutions and according to the obtained validation parameters, results encourage the use of the proposed method described for the assay of CFT and CFZ in their pharmaceutical dosage forms. This method can be used successfully without prior separation of the excipients. Chromatograms obtained from pharmaceutical dosage form samples (with IS) are shown in Fig. 5. The results obtained from the analysis of dosage forms are summarized in Table 6. The quantities found were in conformity with the values claimed by the manufacturers.

Chromatogram obtained from flacon dosage forms a Ceftazidime (1) (Fortum®), Cefixime (IS); and b Ceftizoxime (1) (Cefizox®), Cefixime (IS). Experimental conditions as in chromatographic procedure. The chromatograms are monitored at 260 nm (A) and 290 nm (B)
Recovery experiments were realized using by the standard addition method. Recovery experiments using the developed assay procedure further indicated the absence of interference from commonly encountered pharmaceutical excipients used in the selected formulation (Table 6). Hence, the accuracy was determined using as the difference between nominal and measured concentration (% Bias). It is concluded that the method is sufficiently accurate and precise in order to be applied to flacon dosage forms.
Conclusion
The extensive study of cephalosporines led us to a conclusion that good knowledge of the pK a values is of great importance both for establishing of the appropriate method for its determination and understanding of the determination process. In this study, the pK a values of CFT and CFZ have been determined in water and MeOH–water binary mixtures (20% v/v) by UV spectrophotometric and LC methods, respectively. This work presents the first study dealing with the determination of pK a values of CFT and CFZ by RP-LC method in MeOH–water binary mixture.
Using the chromatographic system presented here, the separation of studied compounds has been performed. This was realized by the careful selection of the column, which decreased the negative interactions between the stationary phase and the charged species that can lead to peak broadening. The reliability of the RP-LC methodology was investigated statistically. There was no significant difference for the assay tested within day and between days. The method was shown to be linear, sensible, accurate, and reproducible over the range of analysis and it can be used to pharmaceutical formulations containing a single active ingredient within a short analysis time (<12 min), without either interfering in the detection or quantitation of the other inactive ingredients or possible interferences. It provides a fast, accurate and robust method for rapid screening of ceftazidime, ceftizoxime, cefixime and can be easily reproduced by a variety of laboratories.
References
BC Centre for Disease Control Knowledgebase (2008) Annual summary of antibiotics utilization. http://www.bccdc.ca/NR/rdonlyres/B90F0301-8BA5-4C52-907F-017F0C0B2DD5/0/Cephalosporins.pdf. Accessed 1 Feb 2011
Thompson RL (1987) Mayo Clin Proc 62:821–834
Sanders CC, Sanders WE (1986) J Infect Dis 154:792–800
Bethesda MD (1989) American hospital formulary service: drug information 89. American Society of Hospital Pharmacists, pp 119–123
Balant L, Dayer P, Auckenthaler R (1985) Clin Pharmacokinet 10:101–143
Gluck SJ, Cleveland JA Jr (1994) J Chromatogr A 680:43–48
Mrestani Y, Reinhard N, Angela M, Michael W (1998) J Chromatogr A 803:273–278
Poole CF, Poole SK (1991) Chromatography today. Elsevier, Amsterdam
Schoenmakers PJ, Tijsen R (1993) J Chromatogr A 656:577–590
Albert A, Sergeant EP (1984) Ionization constants of acids and bases. Chapman and Hall, London
Demiralay EC, Alsancak G, Ozkan SA (2009) J Sep Sci 32:2928–2936
Gumustas M, Sanli S, Sanli N, Ozkan SA (2010) Talanta 82:1528–1537
Mitchell RC, Salter CJ, Tam KY (1999) J Pharm Biomed Anal 20:289–295
Hardcastle JE, Jano I (1998) J Chromatogr B 717:39–56
Şanlı S, Şanlı N, Alsancak G (2009) J Braz Chem Soc 20:939–946
Şanlı S, Altun Y, Şanlı N, Alsancak G, Beltran J (2009) J Chem Eng Data 54:3014–3021
Nahata MC, Morosco RS (1992) J Liq Chromatogr 15:1507–1511
Pehourcq F, Jarry C (1998) J Chromatogr A 812:159–178
Barnes AR (1995) J Liq Chromatogr 18:3117–3128
Abdel-Hamid ME (1998) Il Farmaco 53:132–138
Moore CM, Sato K, Katsumata Y (1991) J Chromatogr A 539:215–220
Williams D (2002) pK a values for some drugs and miscellaneous organic acids and bases. In: Williams DA, Lemke TL (ed) Foye’s principles of medicinal chemistry, edn 5, Lippincott Williams and Wilkins, Philadelphia, pp 1071
El-Shaboury Salwa RA, Saleh Gamal A, Mohamed Fardous A, Rageh Azza H (2007) J Pharm Biomed Anal 45:1–19
Evagelou V, Tsantili-Kakoulidou A, Koupparis M (2003) J Pharm Biomed Anal 31:1119–1128
Aleksic M, Savic V, Popovic G, Buric N, Kapetanovic V (2005) J Pharm Biomed Anal 39:752–756
Rondinini S, Mussini PR, Mussini T (1987) Pure Appl Chem 59:1549–1560
Mussini PR, Mussini T, Rondinini S (1997) Pure Appl Chem 69:1007–1014
Marcus Y (1989) J Chem Soc Faraday Trans 85:381–388
Gran G (1952) Analyst 7:661–671
Beltran JL, Codony R, Prat MD (1993) Anal Chim Acta 276:441–454
Sherrod PH (2010) NLREG, v.4.0. http://www.sandh.com/Sherrod
Pistos CM, Tsantili-Kakoulidou A, Koupparis M (2003) J Chrom Rel Tech 26:937–952
Kasai M, Hatano S, Kitagawa M, Yoshimi A, Nishimura K, Mori N, Sakai A, Sugihara T (1999) Chem Pharm Bull 47:1081–1088
Riley CM, Rosanske TW (1996) Development and validation of analytical methods. Elsevier, New York
Swartz ME, Krull IS (1997) Analytical development and validation. Marcel Dekker, New York
Acknowledgments
We gratefully acknowledge to Dr. Jose Luis Beltran from University of Barcelona for kindly providing the NLREG 4.0 and STAR programs.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sanli, S., Sanli, N., Gumustas, M. et al. Simultaneous Estimation of Ceftazidime and Ceftizoxime in Pharmaceutical Formulations by HPLC Method. Chromatographia 74, 549–558 (2011). https://doi.org/10.1007/s10337-011-2116-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-011-2116-1