
Abstract
Phytophthora cactorum, P. cinnamomi and P. lateralis were reported to be pathogenic on kiwifruit trees in the main planting areas of China. We attempted to simultaneously detect the three pathogens using a multiplex polymerase chain reaction (PCR) and to survey their occurrence in the main production areas. Because of the need to combine different primer pairs for the multiplex PCR and the low specificity of published specific primers for P. cactorum, P. cinnamomi and P. lateralis, new species-specific primers for the three species were designed based on the ras-related protein gene, Ypt1. The specificity of the designed primers was demonstrated using 52 isolates, including 44 Phytophthora species, three Pythium species, and three other soil-borne pathogens. A multiplex PCR method for the simultaneous detection of P. cactorum, P. cinnamomi and P. lateralis was established, and the three pathogens were detected in artificially and naturally infested soils, indicating that these markers can be used in the diagnosis of kiwifruit Phytophthora diseases. In a survey of these pathogens in the main kiwifruit planting areas of China, 99 soil samples were collected at different locations and in different seasons and subjected to the new method, and the distribution of the three pathogens in the main kiwifruit planting areas of China was determined.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Phytophthora, which includes some of the most destructive plant pathogens, causes considerable economic losses to food crops and ornamentals (O’Brien et al. 2009). Several Phytophthora species, including P. cryptogea, P. citrophthora, P. drechsleri, P. palmivora, P. cactorum, P. cinnamomi, P. megasperma, P. citricola and P. lateralis, have been reported to be associated with kiwifruit diseases (Akilli et al. 2011; Baudry et al. 1991; Conn et al. 1991; Kurbetli and Ozan 2013; Latorre et al. 1991; Lee et al. 2001; Mahdavi 2013; Stewart and McCarrison 1991).
Over the past 30 years, kiwifruit (Actinidia chinensis and A. deliciosa) cultivation has dramatically increased in China, which has produced the most kiwis and has the largest area planted with kiwis in the world since 2013 (Ferguson 2014). Shaanxi Province alone produces at least 600,000 tons of kiwifruit each year, almost half of the total for China and more than any other country (Ferguson 2014).
Although Phytophthora cactorum, P. cinnamomi, and P. lateralis were first isolated from diseased kiwifruit roots in Henan Province in the 1990s (Huang and Qi 1998; Wang and Cao 1999), no other investigation of their distribution across China has been conducted in the last 20 years, even though root rot of kiwifruit trees is one of the most serious diseases of kiwifruit in China. Therefore, it is important to develop a simple and rapid method to detect and distinguish these pathogens.
It is often difficult to control diseases caused by Phytophthora spp. because the pathogens release resistant perennating oospores or chlamydospores into the soil. Early detection and diagnosis of the pathogen in plants, soil, or water is thus essential for effective disease control. Conventional and real-time PCR have emerged as important tools for the diagnosis and study of Phytophthora species and have solved some of the problems associated with their detection, control and containment (Kostov et al. 2016; Martin et al. 2012; O’Brien et al. 2009; Schena et al. 2008). Moreover, diagnostic PCR methods and specific primers have been developed for Phytophthora species, including P. cactorum (Bhat and Browne 2010; Causin et al. 2005; Li et al. 2011; Schena et al. 2008), P. cinnamomi (Engelbrecht et al. 2013; Kong et al. 2003; Langrell et al. 2011; O’Brien 2008; Williams et al. 2009), and P. lateralis (Schena et al. 2008; Schenck et al. 2016; Winton and Hansen 2001); however, most of these studies targeted the detection of a single pathogen. Moreover, because some closely related Phytophthora spp. were not distinguished in previous studies, it remains difficult to establish whether those primers are actually specific (Kunadiya et al. 2017; Li et al. 2011).
Multiplex PCR assays allow the simultaneous detection of several species and facilitate large-scale sample processing (Cooke et al. 2007; Martin et al. 2012; O’Brien et al. 2009); however, multiplex PCR has rarely been applied in plant pathology (Bilodeau et al. 2014; Li et al. 2011, 2013), partially because of difficulties in developing multiplex assays and the lower sensitivity of multiplex PCR compared with simplex PCR (Li et al. 2013; Schena et al. 2006). Recently, some new PCR detection techniques, such as the loop-mediated isothermal amplification (LAMP) assay (Feng et al. 2015, 2018; Hansen et al. 2016; Khan et al. 2017, Si Ammour et al. 2017) and the isothermal RPA (recombinase polymerase amplification) assay (Miles et al. 2014; Rojas et al. 2017) were developed for on-site detection of Phytophthora species. However, these reactions are easily contaminated, and the techniques are expensive and incapable of multi-target detection.
Most of these PCR-based diagnostics (PCRDs) are based on the internal transcribed spacer (ITS) regions of nuclear-encoded ribosomal DNA (rDNA) genes or sequence characterized amplified regions (SCAR) (O’Brien et al. 2009). However, ITS sequences are not always sufficiently variable to distinguish closely related taxa (Kroon et al. 2004; Li et al. 2011; Schena et al. 2006), and the development of SCAR primers is very laborious (Schena et al. 2004). Therefore, recently reported studies focused on the cox 1 and cox 2 genes of mitochondrial DNA, as well as the β-tubulin gene, elicitin gene, and ras-related protein gene Ypt1 (Chen and Roxby 1996) in some Phytophthora species (Martin et al. 2012; Meng and Wang 2010; Schena et al. 2006, 2008). Among alternative target genes proposed as the basis of PCRDs, the ras-related protein gene Ypt1 possesses conserved exons and highly variable introns suitable for the development of PCRDs for almost all Phytophthora species (Schena and Cooke 2006).
In this study, we developed a multiplex PCR assay for the simultaneous detection of P. cactorum, P. cinnamomi, and P. lateralis, all of which infect kiwifruit plants in China. For the PCR, we designed primers specific for P. cactorum, P. cinnamomi, and P. lateralis based on the Ypt1 gene. We then successfully applied the novel multiplex PCR technique to samples from infested fields to investigate the distribution of the three pathogens in the main kiwifruit cultivation regions of China.
Materials and methods
Species and strain maintenance
A total of 44 Phytophthora spp. and six additional common soil-borne pathogens including Pythium, Fusarium, Rhizoctonia, and Verticillium spp. were used in this study (Table 1). Fifty-two isolates containing 17 type culture isolates were provided by several scientific resource institutions, including the CBS (Centraalbureau fur Schimmelcultures, The Netherlands), the WPC (World Phytophthora Genetic Resource Collection, USA), the MAFF (Ministry of Agriculture, Forestry and Fisheries, Japan), the NBRC (NITE Biological Resource Centre, Japan) and Gifu University of Japan. Other local isolates were collected from kiwifruit planting fields in Shaanxi Province, China. The Phytophthora spp. and all culturable isolates were maintained on corn meal agar or potato dextrose agar at 20 °C in the dark.
Primer design
The Ypt1 gene sequences from the 50 isolates of Phytophthora species and three of Pythium (Table 2) were aligned to develop specific primers for P. cactorum, P. cinnamomi and P. lateralis by BioEdit ver. 7.0.0 [Ionis (formerly Isis) Pharmaceuticals, Dublin, Ireland]. All Ypt1 gene sequences were collected from the National Center for Biotechnology Information (NCBI) DNA database. Candidate primers were analyzed for dimer and hairpin loop structures (Primer Premier Ver. 5.0; Premier Biosoft International, Palo Alto, CA, USA).
Sequencing
To amplify Ypt1 genes of Phytophthora spp., the Phytophthora universal primers Yph1F_mod2 and Yph2R_mod2 (Table 3) were designed and used in this study. The primer set (Yph1F_mod2/Yph2R_mod2) was a modified version based on the Phytophthora genus-specific primer set (Yph1F/Yph2R) reported by Schena et al. (2008). And the applicability of the modified primers was verified with 108 different Phytophthora species (data not shown). The reaction mixture contained 1 µM each primer, 1 U of Takara Taq DNA polymerase (Takara Bio, Shiga, Japan), 0.2 mM dNTP mixture, 1× PCR buffer [10 mM Tris–HCl (pH 8.3), 50 mM KCl, and 1.5 mM MgCl2], 10 ng of bovine serum albumin (Sigma-Aldrich, St. Louis, MO, USA), and about 50 ng of DNA template in a total volume of 25 µl. PCR was conducted in a DNA thermal cycler (Gene Amp PCR System 2700; Applied Biosystems, Foster City, CA, USA) under the following conditions: 95 °C for 2 min; 40 cycles of denaturation at 94 °C for 30 s, annealing at 62 °C for 45 s, and extension at 72 °C for 30 s; and a final extension at 72 °C for 10 min. After purification of the PCR products, a BigDye Terminator ver. 3.1 Cycle Sequencing kit (Thermo Fisher Scientific, Waltham, MA, USA) was used for cycle sequencing on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). Consensus sequences were generated based on the forward and reverse sequences using Chromas Pro (ver. 1.33; Technelysium Pty. Ltd., Tewantin, Australia).
Collection of soil samples
A total of 99 soil samples in two main kiwifruit production areas (Zhouzhi and Meixian Prefectures) in Shaanxi Province of China were collected in April, July and October of 2017 to survey the pathogen distribution (Table 4). In Zhouzhi Prefecture, 51 soil samples were collected from 10 orchards. In Meixian Prefecture, 48 soil samples were collected from 9 orchards. In each orchard, 5–6 soil samples were collected. For each soil sample, four subsamples (each about 100 g) were randomly collected and mixed thoroughly. From the soil mixture, approximately 200 g was removed and stored at 5 °C. The soil pH was then measured with a FieldScout pH 400 m (Spectrum Technologies, Aurora, IL, USA). Soil texture was determined according to the criterion of the International Society of Soil Science (Li et al. 2011). Disease histories of the kiwifruit planting fields were provided by local agricultural research centers.
DNA extraction from mycelia and soil
Total genomic DNA from mycelia was extracted as described by Kageyama et al. (2003). Mycelia grown on V8 juice broth were used for DNA extraction from culturable species. For soil DNA extraction, the method refined by Kageyama et al. (2003) was modified by incorporating a magnetic bead purification step (MagExtractor-Plant Genome; Toyobo Co., Osaka, Japan) to purify soil DNA extracts as described by Li et al. (2010). Briefly, 0.2 g of soil was added to autoclaved 2.0-ml Eppendorf tubes containing 0.2 g of 1-mm-diameter glass beads. The soil was then suspended in 250 µl of extraction buffer [100 mM Tris HCl (pH 9.0), 40 mM EDTA, 2% (wt/vol) sodium dodecyl sulfate, 0.8% [wt/vol] skim milk; Difco Laboratories, Detroit, MI, USA), and RNase A at 200 µg/ml (Nippongene, Toyama, Japan), then vigorously vortexed at 4200 rpm for 1 min. Next, benzyl chloride (150 µl) was added to the mixture, and the tube was again vigorously vortexed for 2 min. The samples were then incubated at 60 °C for 15 min, after which 150 µl of 3 M sodium acetate was added to the suspension, and the mixture was lightly vortexed. After 15 min of incubation on ice, this suspension was cleared by two rounds of centrifugation at 18,000×g for 10 min, and the upper layer was transferred to a clean tube. Purification of the extracted DNA was subsequently performed according to the manufacturer’s instructions in the purification step of the MagExtractor-Plant Genome kit (Toyobo Co., Osaka, Japan). Finally, 50 µl of the purified DNA was obtained.
Simplex PCR with species-specific primers
The specificity of each developed primer pair was confirmed by simplex PCR with the isolates listed in Table 1. The reaction mixture contained 0.5 µM developed primers, 0.675 U of Takara Taq Hot Start Ver. DNA polymerase (Takara Bio,), 0.2 mM dNTP mixture, 1× PCR buffer [10 mM Tris–HCl (pH 8.3), 50 mM KCl, and 2.0 mM MgCl2], 10 ng of bovine serum albumin (Sigma-Aldrich, St. Louis, MO, USA), and about 10 ng of DNA template in a total volume of 25 µl. PCR was conducted in a DNA thermal cycler (Gene Amp PCR System 2700; Applied Biosystems) by subjecting the samples to 95 °C for 2 min; 40 cycles of denaturation at 94 °C for 30 s, annealing at 63 °C for 35 s, and extension at 72 °C for 30 s; and final extension at 72 °C for 10 min. Amplification was confirmed by electrophoresis in 2% certified agarose S (Nippon Gene Co., Tokyo, Japan). Gels were stained with GelRed™ Nucleic Acid Gel Stain (Biotium, Fremont, CA, USA) and photographed under ultraviolet light.
Multiplex PCR
As a positive control to ensure the success of DNA extraction, two fungal universal primers, 18S-69F and 18S-1118R (Table 3), were added to the multiplex PCR reaction mixture. The multiplex PCR reaction mixture was prepared as described for simplex PCR, except that the primers were used at different concentrations as follows: 0.4 µM for primer pairs 18S-69F/18S-1118R, 0.2 µM for P. cactorum specific Yph_cac_R5, 1 µM for P. cinnamomi specific Yph_cin_R2 and P. lateralis specific Yph_lat_R4, and 2.2 µM for commonly shared forward Phytophthora universal primer Yph1F_mod2. Reaction conditions were the same as in the simplex PCR, except that the annealing temperature was 66 °C.
The applicability of the multiplex PCR was tested using soil samples from four kiwifruit-planting fields with Phytophthora disease histories and two artificially infested soil samples. Pure culture DNA mixtures of the three species served as a positive control.
Sensitivity of detection
Sensitivity to P. cactorum, P. cinnamomi, and P. lateralis DNA was tested in both simplex PCR and multiplex PCR. DNA from two P. cactorum isolates (CH989A11 and ZZ017), two P. cinnamomi isolates (WPC2160 and ZZ029) and one P. lateralis isolate (WPC3361) was tested at 10-fold dilutions (250 pg to 25 fg) in the simplex PCR. Two DNA mixtures (CH989A11 × WPC2160 × WPC3361 and ZZ017 × ZZ029 × WPC3361) were diluted from 250 pg to 250 fg for use in the multiplex PCR.
Inoculum addition to soil samples
For producing Phytophthora mycelia to mix with soil samples, a block (about 7 mm in diameter) of a 5-day-old culture of Phytophthora from V8 juice agar was transferred to V8 broth and cultured at 20 °C for 7 days. Mycelia were collected by centrifugation at 4000 rpm for 10 min. The supernatant was discarded, and 100 ml of sterilized distilled water was added to the tube for homogenization at 3000 rpm for 5 min. This mycelial suspension was then mixed thoroughly into 500 g sterilized soil.
Comparison of different DNA polymerases
DNA polymerase is an important factor affecting sensitivity of the PCR detection system. In this study, 10 DNA polymerases including Takara Taq Hot Start Ver., Takara Taq, Takara Ex Taq Hot start Ver., Takara Mighty Amp, Tks Gflex, PrimeSTAR GXL (6 different DNA polymerases from Takara Bio, Inc., Japan), Platinu SuperFi (Thermo Fisher Scientific, USA), FastStart Taq (Roche Applied Science, Germany), KOD FX Neo (Toyobo, Osaka, Japan) and Kaneka High-Speed (Kaneka Corp., Japan) were tested and compared for amplification efficiencies. Takara Taq Hot Start Ver. yielded the highest amplification efficiency (data not shown).
Results
Primer design and specificity tests
The species-specific DNA regions in the Ypt1 gene were sought for P. cactorum, P. cinnamomi, and P. lateralis. Based on variations in Ypt1, species-specific primers (Table 3) for each pathogen were designed. Two universal primers for the genus Phytophthora, Yph1F_mod2 and Yph2R_mod2, were also designed (Table 3). For the reverse species-specific primers (Yph_cac_R5, Yph_cin_R1, Yph_cin_R2, Yph_lat_R2, Yph_lat_R4), the forward Phytophthora universal primer, Yph1F_mod2, was used as the forward primer in PCR. Similarly, for the forward species-specific primer (Yph_lat_F1), the reverse Phytophthora universal primer, Yph2R_mod2, was used as the reverse primer in PCR.
Overall, 52 isolates including 44 Phytophthora spp. together with six soil-borne pathogens (Table 1) were used to test the specificity of the designed primers for P. cactorum, P. cinnamomi, and P. lateralis. The presence of amplified DNA from all isolates was confirmed using the fungal universal primers 18S-69F and 18S-1118R. Primers Yph1F_mod2 and Yph_cac_R5 only amplified a specific band of 112 bp from P. cactorum isolates (Table 1; Fig. 1a), primers Yph1F_mod2 and Yph_cin_R2 amplified a unique band of 176 bp from P. cinnamomi isolates (Table 1; Fig. 2b) and primers Yph1F_mod2 and Yph_lat_R4 amplified unique band of 225 bp from P. lateralis isolates (Table 1; Fig. 2c). Primer pair Yph1F_mod2/Yph_cin_R1 amplified a unique band of 229 bp from P. cinnamomi isolates (data not shown), and for P. lateralis isolates, primer pair Yph_lat_F1/Yph2R_mod2 amplified a 307 bp band and Yph1F_mod2/Yph_lat_R2 amplified a 189 bp band (data not shown).

Specificity tests of specific primers designed for three Phytophthora species based on closely related species. aP. cactorum specific primer pair Yph1F_mod2/Yph_cac_R5; lanes: 1, P. cactorum CH989A11; 2, P. cactorum ZZ017; 3, P. hedraiandra CBS111725; 4, P. idaei WPC6767; 5, P. pseudotsugae WPC10339; 6, P. clandestina WPC3942; 7, P. iranica CBS374.72; 8, P. tentaculata CBS552.96), 9, P. infestans MAFF305586; 10, P. ipomoeae WPC10225; 11 P. mirabilis WPC3005; 12, P. phaseoli WPC10145; 13, P. nicotianae GF468. bP. cinnamomi specific primer pair Yph1F_mod2/Yph_cin_R2; lanes: 1, P. cinnamomi WPC2160; 2, P. cinnamomi ZZ029; 3, P. parvispora CBS411.96; 4, P. sp. niederhauserii CH96HE1; 5, P. sojae WPC7358; 6, P. pistaciae CBS137185; 7, P. melonis WPC6870; 8, P. cajani WPC3105; 9, P. vignae Ph-9; 10, P. uliginosa CBS109054; 11, P. europaea CBS109049; 12, P. fragariae CBS209.46; 13, P. cambivora WPC0592. cP. lateralis specific primer pair Yph1F_mod2/Yph_lat_R4; lanes: 1 and 2, P. lateralis WPC3361; 3, P. ramorum CBS101553; 4, P. hibernalis CBS114104; 5, P. foliorum WPC10974; 6, P. syringae MAFF645010; 7, P. primulae CBS620.97; 8, P. brassicae CBS179.87; 9, P. sansomeana WPC3163; 10, P. medicaginis WPC10138; 11, P. drechsleri WPC1087; 12, P. cryptogea WPC1088. N sterile distilled water. Size marker: 100-bp DNA ladder

Sensitivity tests using specific primers designed for Phytophthora cactorum, P. cinnamomi and P. lateralis in single-plex PCR with 250 pg to 25 fg DNA from pure cultures of tested isolates or 250 pg to 250 fg in multiplex PCR. Amplification with species-specific primer pair a Yph1F_mod2/Yph_cac_R5 for P. cactorum isolate CH989A11, b Yph1F_mod2/Yph_cin_R2 for P. cinnamomi isolate WPC2160, c Yph1F_mod2/Yph_lat_R4 for P. lateralis isolate WPC3361. d Multiplex PCR with four primers (Yph1F_mod2, Yph_cac_R5, Yph_cin_R2 and Yph_lat_R4). N sterile distilled water. Size marker: 100-bp DNA ladder
Sensitivity tests
In the simplex PCR, primers Yph1F_mod2 and Yph_cac_R5 detected as little as 250 fg of DNA of P. cactorum isolate CH989A11 (Fig. 2a). The same sensitivity was also obtained for P. cactorum isolate ZZ017. Specific primer pairs Yph1F_mod2/Yph_cin_R2 and Yph1F_mod2/Yph_lat_R4 were sensitive to 250 fg of DNA of P. cinnamomi isolate WPC2160 and P. lateralis isolate WPC3361, respectively (Fig. 2b, c). The same sensitivity was also obtained for P. cinnamomi isolate ZZ029 using the primer pair Yph1F_mod2/Yph_cin_R2. For multiplex PCR, the sensitivity was 2.5 pg of DNA for each species from two DNA mixtures: CH989A11 × WPC2160 × WPC3361 (Fig. 2d) and ZZ017 × ZZ029 × WPC3361.
Application of multiplex PCR in naturally and artificially infested soils
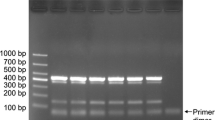
In the multiplex PCR, four species-specific primers (Yph1F_mod2, Yph_cac_R5, Yph_cin_R2 and Yph_lat_R4) together with two fungal universal primers (18S-69F and 18S-1118R) were used. Multiplex PCR analysis of naturally and artificially infested soils amplified fragments obtained from all samples using the fungal universal primers 18S-69F/18S-1118R (Fig. 3), and a specific fragment was amplified, respectively, from samples infested with P. cactorum and P. cinnamomi.

Multiplex PCR from artificially and naturally infested soils to detect Phytophthora cactorum, P. cinnamomi and P. lateralis. Soil sample 1 was artificially infested with P. cactorum and P. lateralis. Soil sample 2 was artificially infested with P. cinnamomi and P. lateralis. Soil samples 3–6 were collected from the main kiwifruit areas in Shaanxi Province of China. P mixture of mycelial DNA from P. cactorum, P. cinnamomi, and P. lateralis as a positive control, N sterile distilled water as a negative control. Size marker: 100-bp DNA ladder
Detection of P. cactorum, P. cinnamomi and P. lateralis in the main kiwifruit planting areas of China
Although the soil samples showed different soil textures (Table 4), the fungal universal primers 18S-69F and 18S-1118R amplified DNA from all samples, indicating successful extraction and amplification. The Phytophthora universal primers were used before the multiplex PCR to detect Phytophthora spp. with the same sensitivity level to the simplex PCR. No pathogens were detected in the 15 samples collected from Zhouzhi Prefecture in April. Among the 20 samples collected in July, P. cactorum was detected in two samples from a field with Phytophthora rot history, and P. cinnamomi was detected in one sample with root rot history. Among the 16 samples collected in October, P. cactorum was detected in two with leaf yellowing history. In Meixian Prefecture, no pathogens were detected in samples collected in April and October, only P. cactorum was detected in three samples with root rot history in July, and P. lateralis was not found in any samples.
In summary, P. cactorum and P. cinnamomi were found in Zhouzhi Prefecture, only P. cactorum was found in Meixian Prefecture, and P. lateralis was not found in either prefecture.
Discussion
In this study, we developed a reliable method to identify and detect P. cactorum, P. cinnamomi and P. lateralis simultaneously. New species-specific primers were designed based on the Ypt1 gene for the three species, then the multiplex PCR was optimized and successfully applied to survey all three pathogens in soils.
Primer specificity is crucial for PCR-based diagnosis. In preliminary tests, we examined nine DNA loci, including the rDNA ITS region, 28S rDNA, 60S ribosomal protein L10 gene, ß-tubulin gene, elongation factor 1 α gene, enolase gene, heat shock protein 90 gene, tigA gene fusion protein sequence, and the Ypt1 gene. When the sequences of species belonging to Phytophthora clade 1 (described by Blair et al. 2008), which is closely related to P. cactorum, were compared to identify interspecies variations suitable for the definition of specific primers, only the Ypt1 gene emerged as a promising candidate for P. cactorum. Li et al. (2011) also designed two specific primers for P. cactorum based on the Ypt1 and compared it with most of the reported specific primers for P. cactorum; it was the most reliable for distinguishing P. cactorum from other closely related species (P. hedraiandra, P. idaei, and P. pseudotsugae).
Several species-specific primers for P. cinnamomi and P. lateralis were published in recent studies (Engelbrecht et al. 2013; Kong et al. 2003; Langrell et al. 2011; Miles et al. 2014; O’Brien 2008; Schena et al. 2008; Schenck et al. 2016; Williams et al. 2009; Winton and Hansen 2001). However, no specificity tests against closely related species have been conducted for most of the species-specific primers that have been published. In our experience, specificity of these published species-specific primers should be reconfirmed against closely related species in the same clade before use. Our opinion is supported by the results of a study by Kunadiya et al. (2017), who tested eight sets of different species-specific primers reported by different researchers for P. cinnamomi against closely related species from clade 7 and found that only three sets were truly specific to P. cinnamomi. The number of known Phytophthora species has doubled in the past 20 years; therefore, it is difficult to obtain cultures or DNA for all species. Nevertheless, researchers should at least attempt to obtain and test species belonging to same phylogenetic clade or sub-clade.
The ITS regions of rDNA are useful targets for fungal species-specific primers because of their high copy number, sequence variability, and fidelity among pathogen species or subspecies. Therefore, they have been widely applied to identify and detect Phytophthora spp. in recent years. However, sequence variations in this region between closely related species are not always sufficient to define highly specific primers. Another disadvantage for rDNA genes is the intraspecific variation of copy number (Meng and Wang 2010; Spies et al. 2011), which may have an effect on the accuracy of pathogen quantification. Moreover, variations in a target locus at an intraspecific level may also limit the utility for analysis of community structure (Lamour 2013).
Unlike rDNA genes, which are generally present in multiple copies, Ypt1 is present as a single copy. In sensitivity tests, DNA of P. cactorum, P. cinnamomi, and P. lateralis was detected at levels as low as 250 fg by simplex PCR analysis (Fig. 1). Although the Ypt1 gene is inferior to rDNA genes with respect to sensitivity, this level of sensitivity appears sufficient for detection and quantification. Schena et al. (2008) used a nested approach based on a first-round amplification with Phytophthora-genus-specific primers and a second amplification with Phytophthora-species-specific multiplex real-time PCR and found that sensitivity increased to 100 fg. However, in the present study, a comparable level of sensitivity was obtained in a single round of amplification.
For most practical applications, the lower level of sensitivity achieved with Ypt1 might be a minor problem; however, the fact that the gene exists in a single copy suggests that single propagules of target species could be detected by a single multiplex real-time PCR. Methods based on single-copy genes are not affected by the number of repeats as in multi-copy genes, and there is the potential to correlate Ct values accurately with the pathogen biomass and/or the number of propagules (Li et al. 2013). Therefore, for application in pathogen quantification, primers designed for Ypt1 should be more suitable than those from multi-copy genes.
Multiplex PCR was successfully performed using the fungal universal primers (18S-69F and 18S-1118R) and species-specific primers (Yph1F_mod2, Yph_cac_R5, Yph_cin_R2, and Yph_lat_R4). According to the critical parameters in multiplex PCR discussed by Henegariu et al. (1997), we optimized four factors: annealing temperature (AT), buffer concentration, primer amounts, and the balance of dNTP and magnesium chloride. When we designed the species-specific primers for the three pathogens based on Ypt1, we tested the Phytophthora universal primers as common forward or reverse primers for the three species to reduce primer amounts as far as possible. Therefore, we re-designed new species-specific primers, rather than using published primers. Moreover, to balance the competitive reaction for Ypt1 for the simultaneous amplification of three different targets, we adjusted the concentrations of different species-specific primers.
For investigation of the distribution of P. cactorum, P. cinnamomi, and P. lateralis in two main kiwifruit planting prefectures of China, samples with different soil properties and disease histories were collected. Using the DNA extraction method refined by Kageyama et al. (2003) and modified by incorporating a magnetic bead purification step (Li et al. 2010), we ensured that the DNA extracted was of high quality and sufficient quantity, as further corroborated by pre-amplification with the 18S gene fungal universal primers (Table 3). We found that more kiwifruit trees suffered from bacterial canker, leaf yellowing, or root rot, rather than the diseases caused by Phytophthora spp. in Zhouzhi and Meixian prefectures. A few samples had a known Phytophthora rot history, but P. cactorum was detected in some samples without Phytophthora rot history, indicating that several kiwifruit fields could potentially be infected with Phytophthora spp. In a recent investigation of kiwifruit Phytophthora diseases around the world, major pathogenic species included P. cryptogea, P. citrophthora, P. drechsleri, P. palmivora, P. cactorum, P. cinnamomi, P. megasperma, P. citricola and P. lateralis. However, in the present investigation, only P. cactorum and P. cinnamomi were detected in Zhouzhi and Meixian prefectures of China.
We note that it is difficult to judge contamination in a field using the multiplex PCR alone in practical application because whether the disease will develop or not depends on the amount of Phytophthora spp. and environmental conditions. In addition, even if other Phytophthora spp. are present, it cannot detect them. Therefore, we suggest doing a preliminary selection by using the Phytophthora genus-specific primers (Yph1F_mod2 and Yph2R_mod2), which proved to be equally sensitive as the specific primers (data not shown). Besides, because of the lower detection limit of the multiplex PCR, simplex PCR is suggested when a soil sample yields positive result with the genus-specific primers but is negative in the multiplex PCR.
Kiwifruit vine is commonly infected by pathogenic bacteria, fungi, and viruses. Some symptoms of Phytophthora diseases and diseases caused by other pathogens are very similar, but fungicides used for their control are completely different. Therefore, incorrect diagnoses will cause avoidable problems and financial loss. Although Phytophthora rot has not severely threatened the main kiwifruit production areas in China thus far, it is still helpful to differentiate Phytophthora disease from those caused by other pathogens to prevent serious economic losses. The technique introduced here was useful and effective in discriminating these pathogens and will be helpful in the early diagnosis of seedling infection and disease control.
References
Akilli S, Serçe ÇU, Zekaİ Katircioğlu Y, Karakaya A, Maden S (2011) Involvement of Phytophthora citrophthora in Kiwifruit decline in Turkey. J Phytopathol 159:579–581
Asano T, Senda M, Suga H, Kageyama K (2010) Development of multiplex PCR to detect five Pythium species related to turfgrass diseases. J Phytopathol 158:609–615
Baudry A, Morzieres J, Ellis R (1991) Effect of Phytophthora spp. on kiwifruit in France. N Z J Crop Hortic Sci 19:395–398
Bhat RT, Browne GT (2010) Specific detection of Phytophthora cactorum in diseased strawberry plants using nested polymerase chain reaction. Plant Pathol 59:121–129
Bilodeau GJ, Martin FN, Coffey MD, Blomquist CL (2014) Development of a multiplex assay for genus- and species-specific detection of Phytophthora based on differences in mitochondrial gene order. Phytopathology 104:733–748
Blair JE, Coffey MD, Park S-Y, Geiser DM, Kang S (2008) A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences. Fungal Genet Biol 45:266–277
Causin R, Scopel C, Grendene A, Montecchio L (2005) An improved method for the detection of Phytophthora cactorum (L.C.) Schröeter in infected plant tissues using SCAR markers. J Plant Pathol 87:25–35
Chen Y, Roxby R (1996) Characterization of a Phytophthora infestans gene involved in vesicle transport. Gene 181:89–94
Conn KE, Gubler WD, Mircetich SM, Hasey JK (1991) Pathogenicity and relative virulence of nine Phytophthora spp. from kiwifruit. Phytopathology 81:974–979
Cooke DEL, Schena L, Cacciola SO (2007) Tools to detect, identify and monitor Phytophthora species in natural ecosystems. J Plant Pathol 89:13–28
Engelbrecht J, Duong TA, Van den Berg N (2013) Development of a nested quantitative real-time PCR for detecting Phytophthora cinnamomi in Persea americana rootstocks. Plant Dis 97:1012–1017
Feng W, Ishiguro Y, Hotta K, Watanabe H, Suga H, Kageyama K (2015) Simple detection of Pythium irregulare using loop-mediated isothermal amplification assay. FEMS Microbiol Lett 362:fnv174
Feng W, Nukaya A, Satou M, Fukuta N, Ishiguro Y, Suga H, Kageyama K (2018) Use of LAMP detection to identify potential contamination sources of plant-pathogenic Pythium species in hydroponic culture systems of tomato and eustoma. Plant Dis 102:1357–1364. https://doi.org/10.1094/PDIS-10-17-1679-RE
Ferguson AR (2014) Kiwifruit in the world. Acta Hortic 1096:33–46
Hansen ZR, Knaus BJ, Tabima JF, Press CM, Judelson HS, Grünwald NJ, Smart CD (2016) Loop-mediated isothermal amplification for detection of the tomato and potato late blight pathogen, Phytophthora infestans. J Appl Microbiol 120:1010–1020
Henegariu O, Heerema NA, Dlouhy SR, Vance GH, Vogt PH (1997) Multiplex PCR: critical parameters and step-by-step protocol. BioTechniques 23:504–511
Huang Y, Qi P (1998) A study on the cause of root rot disease of kiwifruit trees in Guangdong Province (in Chinese). J South China Agric Univ 19:19–22
Kageyama K, Komatsu T, Suga H (2003) Refined PCR protocol for detection of plant pathogens in soil. J Gen Plant Pathol 69:153–160
Khan M, Li B, Jiang Y, Weng Q, Chen Q (2017) Evaluation of different PCR-based assays and LAMP method for rapid detection of Phytophthora infestans by targeting the Ypt1 gene. Front Microbiol 8:1920
Kong P, Hong CX, Richardson PA (2003) Rapid detection of Phytophthora cinnamomi using PCR with primers derived from the Lpv putative storage protein genes. Plant Pathol 52:681–693
Kostov K, Verstappen ECP, Bergervoet JHW, de Weerdt M, Schoen CD, Slavov S, Bonants PJM (2016) Multiplex detection and identification of Phytophthora spp. using target-specific primer extension and Luminex xTAG technology. Plant Pathol 65:1008–1021
Kroon LPNM, Bakker FT, van den Bosch GBM, Bonants PJM, Flier WG (2004) Phylogenetic analysis of Phytophthora species based on mitochondrial and nuclear DNA sequences. Fungal Genet Biol 41:766–782
Kunadiya M, White D, Dunstan WA, Hardy GESJ, Andjic V, Burgess TI (2017) Pathways to false-positive diagnoses using molecular genetic detection methods; Phytophthora cinnamomi a case study. FEMS Microbiol Lett 364:fnx009
Kurbetli I, Ozan S (2013) Occurrence of Phytophthora root and stem rot of kiwifruit in Turkey. J Phytopathol 161:887–889
Lamour K (ed) (2013) Phytophthora: a global perspective. CABI, Wallingford
Langrell SRH, Morel O, Robin C (2011) Touchdown nested multiplex PCR detection of Phytophthora cinnamomi and P. cambivora from French and English chestnut grove soils. Fungal Biol 115:672–682
Latorre BA, Alvarez C, Ribeiro OK (1991) Phytophthora root rot of kiwifruit in Chile. Plant Dis 75:949–952
Lee YH, Jee HJ, Cha KH, Ko SJ, Park KB (2001) Occurrence of Phytophthora root rot on kiwifruit in Korea. Plant Pathol J 17:154–158
Li M, Senda M, Komatsu T, Suga H, Kageyama K (2010) Development of real-time PCR technique for the estimation of population density of Pythium intermedium in forest soils. Microbiol Res 165:695–705
Li M, Asano T, Suga H, Kageyama K (2011) A multiplex PCR for the detection of Phytophthora nicotianae and P. cactorum, and a survey of their occurrence in strawberry production areas of Japan. Plant Dis 95:1270–1278
Li M, Inada M, Watanabe H, Suga H, Kageyama K (2013) Simultaneous detection and quantification of Phytophthora nicotianae and P. cactorum, and distribution analyses in strawberry greenhouses by duplex real-time PCR. Microbes Environ 28:195–203
Mahdavi E (2013) Occurrence of Phytophthora root and collar rot disease of kiwifruit orchards in the west part of the Mazandaran Province. Scholarly J Agr Sci 3:331–335
Martin FN, Abad ZG, Balci Y, Ivors K (2012) Identification and detection of Phytophthora: reviewing our progress, identifying our needs. Plant Dis 96:1080–1103
Martin FN, Blair JE, Coffey MD (2014) A combined mitochondrial and nuclear multilocus phylogeny of the genus Phytophthora. Fungal Genet Biol 66:19–32
Meng J, Wang Y (2010) Rapid detection of Phytophthora nicotianae in infected tobacco tissues and soil samples based on its Ypt1 gene. J Phytopathol 158:1–7
Miles TD, Martin FN, Coffey MD (2014) Rapid isothermal detection and species-specific assay of Phytophthora in plant samples using recombinase polymerase amplification (Abstract). Phytopathology 104:80
O’Brien PA (2008) PCR primers for specific detection of Phytophthora cinnamomi. Australas Plant Path 37:69–71
O’Brien PA, Williams N, Hardy GES (2009) Detecting Phytophthora. Crit Rev Microbiol 35:169–181
Rojas JA, Miles TD, Coffey MD, Martin FN, Chilvers MI (2017) Development and application of qPCR and RPA genus- and species-specific detection of Phytophthora sojae and P. sansomeana root rot pathogens of soybean. Plant Dis 101:1171–1181
Schena L, Nigro F, Ippolito A, Gallitelli D (2004) Real-time quantitative PCR: a new technology to detect and study phytopathogenic and antagonistic fungi. Eur J Plant Pathol 110:893–908
Schena L, Hughes KJD, Cooke DEL (2006) Detection and quantification of Phytophthora ramorum, P. kernoviae, P. citricola and P. quercina in symptomatic leaves by multiplex real-time PCR. Mol Plant Pathol 7:365–379
Schena L, Duncan JM, Cooke DEL (2008) Development and application of a PCR-based ‘molecular tool box’ for the identification of Phytophthora species damaging forests and natural ecosystems. Plant Pathol 57:64–75
Schenck N, Fourrier-Jeandel C, Ioos R (2016) A robust and specific real-time PCR tool for the detection of Phytophthora lateralis in plant tissues. Eur J Plant Pathol 146:231–244
Si Ammour M, Bilodeau GJ, Tremblay DM, van der Heyden H, Yaseen T, Varvaro L, Carisse O (2017) Development of real-time isothermal amplification assays for on-site detection of Phytophthora infestans in potato leaves. Plant Dis 101:1269–1277
Spies CFJ, Mazzola M, Botha WJ, Langenhoven SD, Mostert L, McLeod A (2011) Molecular analyses of Pythium irregulare isolates from grapevines in South Africa suggest a single variable species. Fungal Biol 115:1210–1224
Stewart A, McCarrison AM (1991) Excised shoot assay to determine the pathogenicity of root-rotting Phytophthora species on kiwifruit. Australas Plant Path 20:146–148
Wang R, Cao Z (1999) Diagnosis of blight of Actinidia chinensis in Shaanxi Province (in Chinese). J Northwest Agric Forest Univ 27:75–78
Williams N, Hardy GESJ, O’Brien PA (2009) Analysis of the distribution of Phytophthora cinnamomi in soil at a disease site in Western Australia using nested PCR. Forest Pathol 39:95–109
Winton LM, Hansen EM (2001) Molecular diagnosis of Phytophthora lateralis in trees, water, and foliage baits using multiplex polymerase chain reaction. Forest Pathol 31:275–283
Acknowledgements
This research was supported by the National Natural Science Foundation (Grant No. 31500415, 31600445) and the Fundamental Research Funds for the Central Universities (grant nos. GK201703033, GK201703036) of China. We thank Dr. Koji Kageyama of Gifu University for isolates of many important Phytophthora spp. and the Plant Protective Stations of Zhouzhi and Meixian Prefectures for information on soil samples from kiwifruit planting areas.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Research involving human or animal participants
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Bi, X., Hieno, A., Otsubo, K. et al. A multiplex PCR assay for three pathogenic Phytophthora species related to kiwifruit diseases in China. J Gen Plant Pathol 85, 12–22 (2019). https://doi.org/10.1007/s10327-018-0822-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10327-018-0822-3