
Abstract
Mass spectrometry is a major tool for analysing organic pollutants. However, scientists often complain about laborious sample preparation. The development of new commercial high-resolution mass spectrometers gives a chance to improve simultaneously speed, reliability, and sensitivity of the analysis. Here, we used the time-of-flight high-resolution mass spectrometer Pegasus GC-HRT to identify and quantify 55 priority organic pollutants in water samples. This mass spectrometer has a high resolution of 50,000, a high mass accuracy of about 1 ppm and a very high acquisition rate of up to 200 full mass range spectra per second. 1 mL water samples were extracted with 1 mL dichloromethane. Results show that the sample preparation and analysis are achieved 30 times faster, requiring 1,000 times less water and 350 times less solvent than the classic 8270 method of the United States Environmental Protection Agency. The detection limit is 1 μg/L. The quantification limit is 10 μg/L. Our procedure, named accelerated water sample preparation, is simpler, faster, cheaper, safer and more reliable than 8270 Method.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mass spectrometry has demonstrated itself as the most reliable analytical method to identify and quantify organic compounds in environmental samples (Lebedev 2013), while gas chromatography/mass spectrometry (GC/MS) has remained very popular for the targeted and nontargeted analysis of volatile and semivolatile pollutants (Lebedev 2012). The benefits of GC/MS include the universality, sensitivity, reliability, and the availability of mass spectra libraries (NIST 2011). Perhaps the only issue of concern when using GC/MS for the analysis of semivolatile pollutants involves sample preparation. In the majority of the standard methods, the sample preparation procedure is laborious, time-consuming and often brings to certain analytes losses or distorted results. These issues inspired significant efforts to develop faster, easier and more reliable methods of sample preparation. Several recent reviews deal with these modern approaches (Ballesteros-Gomez and Rubio 2011; Zuloaga et al.2012).
The most radical innovation in MS involves ambient ionization methods. The main merits of ambient ionization are simplicity and almost complete absence of sample preparation. Since the first report on desorption electrospray ionization (Takats 2004) in 2004, several dozens of similar methods were developed. Some of them have great perspectives in the environmental sciences (Harris et al. 2011; Huang et al. 2011; Alberici et al. 2012). However, these methods do not involve any efficient separation step, which is a certain drawback when it deals with complex environmental samples with the necessity to detect and quantify many various analytes.
The Quick, Easy, Effective, Rugged, and Safe (QuEChERS) method was developed in 2004 and has been readily accepted by many pesticide residue analysts (Schenck and Hobbs 2004). The method has significantly simplified, improved, and standardized the analysis of pesticides residues in solid phases (e.g., food) by GC/MS and LC/MS.
Membrane inlet mass spectrometry (MIMS) is also quite perspective. It has been applied for the real-time analysis for many years; however, it is not yet widely used. Since MIMS allows avoiding sampling and sample preparation, it is a good alternative to the classic GC/MS methods. The main trends in the development of this technique, especially created for the portative mass spectrometers, are reviewed in (Davey et al. 2011).
Nevertheless, GC/MS method with electron ionization still remains a “workhorse” for the qualitative and quantitative analysis of organic priority pollutants in the environment (Lebedev 2012). The main efforts in improving of this approach are focused on developing more effective, fast, and reliable sample preparation procedures. Different versions of solid-phase extraction (SPE) and solid-phase microextraction (SPME) are quite popular nowadays (Ballesteros-Gomez and Rubio 2011; Zuloaga et al.2012). Various stationary phases for cartridges allow for selective separation of different groups of analytes. As drawbacks of SPE, one can mention the lack of universality and certain increase in the analysis cost.
A simplified version of liquid–liquid extraction on diatomaceous earth in small cartridges was proposed for isolation of organic compounds from 1 mL of water with further MS analysis (Majors 2012). The method is based on adsorption of constituents of aqueous solution on diatomaceous earth with further elution of target organic compounds with an organic solvent. A new tendency in liquid–liquid extraction deals with analyte extraction with minimal amount of solvent. Nowadays, one of the most “trendy” methods is single-drop microextraction. There are many versions of this method. Historically, the first one involved a drop of organic solvent placed on a Teflon rod and introduced into the stirred aquatic sample (Jeannot and Cantwell 1996; Liu and Dasgupta 1996). Later it was modified by two independent groups. A GC syringe was proposed as a tool to sustain a solvent drop (Jeannot and Cantwell 1997; He and Lee 1997). The method has been called “direct immersion.” The use of a syringe as a micro separating column resulted in proposing new technique—dynamic liquid-phase microextraction (He and Lee 1997). In this case, an analyzed water sample is pumped in and out of the syringe containing a solvent (unexposed-drop). In an alternative approach, a solvent drop was maintained at the end of the needle, then sucked into the syringe and then again squeezed to the needle end (exposed-drop). Using an organic solvent with higher boiling point (octanol-1 or hexadecane), it is possible to determine rather efficiently various semivolatile compounds by Headspace SDME (Przyjazny and Kokosa 2002; Theis et al. 2001, Tankeviciute et al. 2001).
It is possible to decrease the volume of water sample as well. Thus, drop-to-drop solvent microextraction is based on several microliters of both sample and organic solvent (Wu et al. 2006). Another method engaged a solvent drop maintained on the syringe needle or on the Teflon tube constantly contacting fresh fluent of sample (Liu and Lee 2000; Chen et al. 2006, Guo et al. 2005; He and Lee 2006). To achieve selective extraction of organic compounds depending on pH, a method of liquid–liquid–liquid microextraction was proposed (Ma and Cantwell 1999; Liu et al. 2012, Zhu et al. 2002). Hollow fiber microextraction is another technique allowing using very small volumes of organic solvents and reasonable volume of aqueous samples (Jiang and Lee 2004). All these methods are fast and do not require large portions of a solvent. However, there are certain technical difficulties of their application.
An interesting approach called liquid–liquid microextraction with solidification of the floating organic was proposed in (Zanjani et al. 2007). The problems with the equilibrium time of this technique were overcome with appearance of dispersive liquid–liquid microextraction (Rezaee et al. 2006). Combination of two latter techniques resulted in dispersive liquid–liquid microextraction with solidification of the organic floating drop (Leong and Huang 2008). This technique demonstrated its applicability to the analysis of quite various organic compounds including polycyclic aromatic hydrocarbons, phthalates, organochlorines, phenols, sulfonamides, etc., with recoveries over 70 % (Vera-Avila et al. 2013).
A fast procedure for GC/MS analysis of perfluorinated compounds in water was proposed recently (Bagheri et al. 2008). Ion-pair reagent (tetrabutylammonium hydrosulfate), a dispersant, and an extracting solvent (chloroform) were added directly to water sample to form extractable ion pairs in conditions of active stirring during 30 s. After 6 min of assertion, the extract was introduced into the injector of gas chromatograph, where tetrabutylammonium salts of perfluorinated acids converted into the corresponding butyl esters. The method is certainly simpler and faster comparing to the earlier alternatives.
Classic 8270 GC/MS method of USA Environmental protection agency (EPA) (US EPA 2007) is widely used all over the world for the detection and quantification of semivolatile priority pollutants in water and other environmental matrixes. Sample preparation of water samples for this method is performed according to 3510C method. It is a standard liquid–liquid extraction in a separating funnel. This procedure is time-consuming (about 3–3.5 h), laborious, requires large volumes of sample (e.g., 1 L of water) and solvent (360 mL of dichloromethane per 1 L of water) and brings to unavoidable losses of analytes, especially while concentrating the extracts. The limits of quantification of all analytes are quite high (10 μg L−1 of water and higher). Besides that the recoveries of analytes with lower boiling points are not spectacular. On the other hand, liquid–liquid extraction with dichloromethane allows extracting the most various classes of semivolatile pollutants which is of primary importance both for the targeted and nontargeted analyses. Therefore, a certain scientific challenge engages improving of liquid–liquid extraction with elimination of the mentioned shortcomings and retaining of its advantages.
Considering that since the development of this method, many years have passed and sensitivity of mass spectrometers have raised significantly, we have attempted to improve the method, making it cheaper, safer, faster, more sensitive and reliable. Actually, using 8270 method with modern mass spectrometers, it is possible to detect targeted and nontargeted analytes at lower levels than that prescribed in the method. This possibility is actively applied all over the world. However, it is as well possible to use an alternative approach involving lower sample volumes with the detection limits for priority pollutants equal to these of the classic 8270 method (US EPA 2007). This is a reasonable idea as these detection limits are satisfactory for the needs of environmental legislation. In our earlier study, we demonstrated such an approach to treat polycyclic aromatic hydrocarbons (PAH) in water by GC/HRMS with accelerated sample preparation (Polyakova et al. 2013).
The present work deals with the application of the earlier proposed method (Polyakova et al. 2013) for the identification and quantification of representatives of several groups of organic pollutants in water samples. Besides PAH phthalates, phenols, organochlorine compounds and some other priority pollutants from the US EPA list (US EPA 2007) were treated. The proposed technique called “accelerated water sample preparation—AWASP” requires GC/HRMS instrument and really small portion of both solvent and water sample. Thus, the sample preparation may be carried out “in the field,” while accurate mass measurements provide the improved reliability of the results.
Materials and methods
Mass spectrometry
All experiments were performed with time-of-flight (TOF) high-resolution mass spectrometer Pegasus GC-HRT (LECO Corporation, Saint Joseph, MI, USA) with Folded Flight Path multiple reflecting geometry of mass analyzer coupled with an Agilent 7890A gas chromatograph (Agilent, Palo Alto, CA, USA). The instrument was capable of acquiring high resolution (up to 50,000 at FWHH in ultra-high-resolution mode) and high mass accuracy (~1 ppm) data at a very high acquisition rate (up to 200 full mass range spectra per second). The system was controlled by the ChromaTOF-HRT software version 1.80 (LECO Corporation), which was also used for data collection and data processing. The data were collected using 10 full (40–300 m/z range) spectra per second in high-resolution mode (25,000 at FWHH). The electron ionization source was kept at the 300 °C. Chromatographic separation was performed using DIOXIN2 (Restek, USA) column (40 m length, internal diameter 180 μm and phase thickness 0.18 μm) with a helium flow of 1 mL min−1; the column temperature was programmed as follows: start at 40 °C—hold for 5 min—ramp at 10 °C min−1 up to 250 °C—then ramp at 6 °C min−1 up to 330 °C—hold 5 min; transfer line temperature—330 °C. 2 μL of sample were introduced into the injector heated at 270 °C in splitless mode; after 75 s, the injector was purged with a flow of helium at 20 mL min−1. The analyte chromatographic peaks were automatically found and quantified using Target Analyte Finding feature of the ChromaTOF-HRT, which uses accurate masses of the molecular ions and certain characteristic fragment ions as one of the criteria for finding and matching analytes.
Chemical standards
Calibration solutions of priority pollutants for all the experiments and solutions of internal standards (perdeuterated naphthalene, acenaphthene, phenanthrene, chrysene, and perylene) were prepared from standard Restek (USA) mixtures. Distilled water used in experiments was HPLC grade, dichloromethane (HPLC grade, ≥99.9 %), and anhydrous granular sodium sulfate (≥99.0 %) produced by Sigma-Aldrich (USA).
Sample preparation
1 mL of water sample was placed in a 5-mL vial, and 1 mL of dichloromethane was added. The sample was vigorously shaken for 1 min. Calculated amount of internal standards (2–100 ng) was added with a syringe to the organic phase, and then, sodium sulfate ~1.5 g was introduced by small portions. The sample was vigorously shaken after each addition. After binding of aqueous phase with sodium sulfate, transparent dichloromethane extract was transferred into the pure vial for further analysis. Then, 2 μL of the extract was injected into GC in splitless mode.
Results and discussion
Taking into account the sensitivity of modern GC/MS systems, it is definitely possible to decrease significantly the volume of an aqueous sample from 1 L prescribed by 8270 EPA method without deteriorating detection limits of the analysis. Since the lowest quantification limits for the semivolatile priority pollutants is 10 μg L−1 (8270 EPA method), 1 mL of water sample should contain not less than 10 ng of each of the target analytes. The declared sensitivity of Pegasus GC-HRT (LECO, USA) mass spectrometer used in the present study exceeds 1 pg. Therefore, even 1 μl of that sample would be enough to detect and quantify reliably all the semivolatile priority pollutants at the required level. Although injection of water sample directly into GC column is possible, it is not a custom as it leads to the rapid deterioration of the column. The required approach should involve quantitative transfer of all the organic constituents from a small water volume into adequate small volume of an organic solvent.
We developed a new accelerated water sample preparation (AWASP) approach. In the present study, 1 mL of water with calculated amounts of priority pollutants (1–100 ng), and 1 mL of dichloromethane as an extraction solvent were used. Anhydrous sodium sulfate as a reagent to bind water simultaneously improved the extraction due to “salting-out” effect. This approach is widely used in classic liquid–liquid extraction for decreasing the solubility of organic compounds in water and increasing their extractability. After addition of all sodium sulfate (~1.5 g), aquatic phase was totally bounded and organic compounds were quantitatively transferred into organic phase. Actually, the process may be considered as a replacement of aquatic phase by organic phase (in this experiment by dichloromethane). All sampling procedures take 5–10 min, and there is no need to concentrate the sample. Therefore, there are no losses of volatile components. Furthermore, the sample preparation could be totally implemented outside the laboratory, in field at any sampling site.
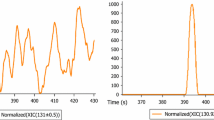
It is worth special mentioning the importance of accurate mass measurements. It allows for better reliability, eliminating false-positive results (Polyakova et al. 2012, 2013) ,and better detection limit, notably reducing effect of background on quantification and simultaneously increasing signal/noise ratio (Polyakova et al. 2013; Lebedev et al. 2013). Nevertheless, the proposed approach may be applied with reasonable results at low-resolution instruments as well. Figure 1 represents the shape of the mass chromatographic peaks observed in the experiments with injection of two polycyclic aromatic hydrocarbons into the column (concentration 10 pg µL−1).

Chromatographic peak shapes in the mass chromatograms used for the quantification of dibenzo[a,h]anthracene (m/z 278.1090, top) and benzo[a]pyrene (m/z 252.0934, bottom). Intensity and shape of the signals allow reliable quantification. The tailing is practically absent
The internal standards’ approach is based on the preliminary calculations of the response factors (RF) for the targeted analytes:
where RF—response factor, M—amount of the compound in the standard solution, S—the signal intensity (peak area); Index is is referred to the internal standard, index x—to the analyte.
Table 1 represents the response factor values for the studied pollutants calculated as averaged results based on triplicate injections at three concentrations (10, 40, and 100 pg/μL). For the majority of PAH, RF values are close to 1, which is the ideal value for the most accurate measurements. The only exception involves methylnaphthalenes (RF ~ 0.5) as the peaks of M+. and [M−H]+ ions of these compounds are of equal intensity.
The recoveries calculated for three concentrations are listed in Table 2. They are compared with the recoveries of the standard US EPA 8270 method (US EPA 2007). As one can see in the case of PAH, the results are quite similar. They are a bit worse for two the heaviest analytes, but much better for the light ones (naphthalene, methylnaphthalenes, and acenaphthylene). The latter issue is very important as it proves the advantage of the proposed approach dealing with the absence of any concentration steps. As a result, these more volatile compounds are not lost during sample preparation. Therefore, the elimination of the concentration stage allows for application of the proposed method to the analysis of volatile compounds (toluene, xylenes, chloroethanes, etc.), combining 8270 and 8260 (partially) EPA methods for semivolatile and volatile compounds in one simpler version.
Same conclusion may be done for rather volatile dichloro- and trichlorobenzenes (Table 2). For these pollutants, AWASP recoveries are about 20 % higher than these values in 8270 EPA method. Table 2 clearly demonstrates that all chlorobenzene congeners are readily amenable for AWASP-GC/HRMS. Similar situation is observed for the light phenols. AWASP recoveries are better than the standard values reported for 8270 method. The worse result for 2,4-dichlorophenol may be rationalized by the fact that before the reported experiments on AWASP, the column and the whole instrument were used for the analysis of polychlorinated dibenzodioxins and their precursors, while this particular phenol is one of them. Reasonable results for phenols are very important, as in 8270 methods, it is necessary to acidify the sample before the extraction (pH 2). Although this stage is omitted in AWASP, the phenols’ extraction takes place readily and almost quantitatively. The results could be improved further using more appropriate chromatographic columns.
There are no problems when it deals with various organochlorines listed in Tables 1 and 2. The recoveries for this group of compounds are always better than in 8270 method. It is worth specially mentioning hexachloroethane which although having high RSD value (20 %) of RF measurements (Table 1) demonstrated excellent recovery (Table 2) due to the elimination of concentration step.
Phthalates AWASP recoveries are slightly worse than in 8270 method, although being acceptable. Very bad result obtained for dibutylphthalate is due to the system contamination with this very widespread pollutant. Speaking about a group of miscellaneous priority pollutants, it is worth emphasizing excellent aniline recoveries achieved in the case of AWASP (~100 vs. 17 %). The cause again involves the absence of the concentration step, which becomes crucial for this quite volatile compound. This result also demonstrates the excellent applicability of the proposed procedure for compounds of both acidic and basic nature.
Another important issue worth special discussion involves the importance of accurate mass measurements. According to the European Commission decision (2002/657/EC) (Eur. Union. 2002), a banned compound is considered “confirmed to be present in the sample” by GC/MS if the retention time is within the acceptance window, and if the detection method provides four identification points. Since measurement of nominal mass provides 1 point, while measurement of accurate mass—2 points, it is obvious that application of HRMS is preferable. Actually, recording of accurate masses for two ions belonging to a certain compound allows considering this compound reliably identified (retention time should be considered as well). One peak (the most intensive) is used for quantification and the second one for the confirmation of identification. Comparing AWASP-GC/HRMS approach with EPA 8270 method, the following issues should be emphasized:
-
1.
Sample preparation time is decreased from 3.5 h to 5–10 min.
-
2.
The required water sample volume is decreased from 1 L to 1 mL.
-
3.
The required dichloromethane volume is decreased from 360 to 1 mL. This fact besides decreasing the cost means that AWASP is safer and “greener”.
-
4.
The requirements for the laboratory equipment and glassware are much lower.
-
5.
It is possible to carry on sample preparation directly on site.
-
6.
It is possible to develop automatic sample preparation methods.
-
7.
It is possible to expand the range of compounds amenable for the analysis including more volatile pollutants.
-
8.
The results of HRMS based on accurate mass measurements are of higher reliability.
-
9.
Due to universality, AWASP approach may be successfully used for the nontargeted GC/MS analysis as well.
Conclusions
The proposed AWASP-GC/HRMS method for the analysis of semivolatile priority pollutants in water samples is cheaper, faster, easier, safer and more reliable than the existing analogs. It is also universal and able to expand the range of analytes, including more volatile species. Sample preparation may be carried out directly on site and may be automated.
References
Alberici RM, Simas RC, Eberlin MN (2012) Ambient mass spectrometry: environmental analysis without sample preparation. In: Lebedev AT (ed) Comprehensive environmental mass spectrometry. ILM Publications, UK, pp 147–166
Bagheri H, Khalilian F, Babanezhad E, Es-haghi A, Rouini MR (2008) Modified solvent microextraction with back extraction combined with liquid chromatography-fluorescence detection for the determination of citalopram in human plasma. Anal Chim Acta 610:211–216
Ballesteros-Gomez A, Rubio S (2011) Recent advances in environmental analysis. Anal Chem 83:4579–4613
Chen X, Zhang T, Liang P, Li Y (2006) Application of continuous-flow liquid phase microextraction to the analysis of phenolic compounds in wastewater samples. Microchim Acta 155:415–420
Davey NG, Krogh ET, Gill CG (2011) Membrane-introduction mass spectrometry (MIMS). Trends Anal Chem 30:1477–1485
Eur. Union. (2002) Commission Decision 2002/657/EC implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off J Eur Union 125(2002):10–32
Guo L, Liang P, Zhang T, Liu Y, Liu S (2005) Use of continuous-flow microextraction and liquid chromatography for determination of phoxim in water samples. Chromatographia 61:523–526
Harris GA, Galhena AS, Fernandez FM (2011) Ambient sampling/ionization mass spectrometry: applications and current trends. Anal Chem 83:4508–4538
He Y, Lee HK (1997) Liquid-phase microextraction in a single drop of organic solvent by using a conventional microsyringe. Anal Chem 69:4634–4640
He Y, Lee HK (2006) Continuous flow microextraction combined with high-performance liquid chromatography for the analysis of pesticides in natural waters. J Chromatogr A 1122:7–12
Huang MZ, Cheng SC, Cho YT, Shiea J (2011) Ambient ionization mass spectrometry: a tutorial. Anal Chim Acta 702:1–15
Jeannot MA, Cantwell FF (1996) Solvent microextraction into a single drop. Anal Chem 68:2236–2240
Jeannot MA, Cantwell FF (1997) Mass transfer characteristics of solvent extraction into a single drop at the tip of a syringe needle. Anal Chem 69:235–239
Jiang X, Lee HK (2004) Solvent bar microextraction. Anal Chem 76:5591–5596
Lebedev AT (2012) Comprehensive environmental mass spectrometry. ILM Publications, UK, p 510
Lebedev AT (2013) Environmental mass spectrometry. Annu Rev Anal Chem 6:163–189
Lebedev AT, Polyakova OV, Mazur DM, Artaev VB (2013) The benefits of high resolution mass spectrometry in environmental analysis. Analyst 138:6946–6953
Leong MI, Huang SD (2008) Dispersive liquid–liquid microextraction method based on solidification of floating organic drop combined with gas chromatography with electron-capture or mass spectrometry detection. J Chromatogr A 1211:8–12
Liu H, Dasgupta PK (1996) Analytical chemistry in a drop. Solvent extraction in a microdrop. Anal Chem 68:1817–1821
Liu W, Lee HK (2000) Continuous-flow microextraction exceeding 1000-fold concentration of dilute analytes. Anal Chem 72:4462–4467
Liu WL, Ko YC, Hwang BH, Li ZG, Yang TCC, Lee MR (2012) Determination of perfluorocarboxylic acids in water by ion-pair dispersive liquid–liquid microextraction and gas chromatography–tandem mass spectrometry with injection port derivatization. Anal Chim Acta 726:28–34
Ma M, Cantwell FF (1999) Chain unfolding in an ODS-bonded phase caused by the sorbed tetra-n-butylammonium ion. Anal Chem 71:388–393
Majors RE (2012) Supported liquid extraction: the best-kept secret in sample preparation. LC GC Eur 25:430–435
Method 8270D Semivolatile organic compounds by gas chromatography/mass spectrometry (GC/MS) (2007) US Environmental Protection Agency
NIST (2011). NIST/EPA/NIH Mass Spectral Database (NIST11). National Institute of Standards and Technology, part of the United States Department of Commerce, Gaithersburg, Maryland, USA. http://chemdata.nist.gov
Polyakova OV, Mazur DM, Bolshov MA, Seregina IF, Lebedev AT (2012) Estimation of contamination of atmosphere of Moscow in winter. J Anal Chem 67:1039–1049 Original Russian version (2012) in Mass-spektrometria (Rus) 9:5–15
Polyakova OV, Mazur DM, Artaev VB, Lebedev AT (2013) Determination of polycyclic aromatic hydrocarbons in water by gas chromatography/mass spectrometry with accelerated sample preparation. J Anal Chem 68:1099–1103 Original Russian version (2012) in Mass-spektrometria (Rus) 9:217–222
Przyjazny A, Kokosa JM (2002) Analytical characteristics of the determination of benzene, toluene, ethylbenzene and xylenes in water by headspace solvent microextraction. J Chromatogr A 977:143–153
Rezaee M, Assadi Y, Hosseini MRM, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid–liquid microextraction. J Chromatogr A 1116:1–9
Schenck FJ, Hobbs JE (2004) Evaluation of the quick, easy, cheap, effective, rugged, and safe (QuEChERS) approach to pesticide residue analysis. Bull Environ Contam Toxicol 73:24–30
Takats Z, Wiseman JM, Gologan B, Cooks RG (2004) Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 306:471–473
Tankeviciute A, Kazlauskas R, Vickackaite V (2001) Headspace extraction of alcohols into a single drop. Analyst 126:1674–1677
Theis AL, Waldack AJ, Hansen SM, Jeannot MA (2001) Headspace solvent microextraction. Anal Chem 73:5651–5654
Vera-Avila LE, Rojo-Portillo T, Covarrubias-Herrero R, Pena-Alvarez A (2013) Capabilities and limitations of dispersive liquid–liquid microextraction with solidification of floating organic drop for the extraction of organic pollutants from water samples. Anal Chim Acta 805:60–69
Wu HF, Yen JH, Chin CC (2006) Combining drop-to-drop solvent microextraction with gas chromatography/mass spectrometry using electronic ionization and self-ion/molecule reaction method to determine methoxyacetophenone isomers in one drop of water. Anal Chem 78:1707–1712
Zanjani MRK, Yamini Y, Shariati S, Jonsson JA (2007) A new liquid-phase microextraction method based on solidification of floating organic drop. Anal Chim Acta 585:286–293
Zhu L, Tay CB, Lee HKJ (2002) Liquid–liquid–liquid microextraction of aromatic amines from water samples combined with high-performance liquid chromatography. J Chromatogr A 963:231–237
Zuloaga O, Navarro P, Bizkarguenaga E, Iparraguirre A, Vallejo A, Olivares M, Prieto A (2012) Overview of extraction, clean-up and detection techniques for the determination of organic pollutants in sewage sludge: a review. Anal Chim Acta 736:7–29
Acknowledgments
The authors would like to acknowledge LECO Corporation for allowing access to the Pegasus GC/HRT and Viatcheslav Artaev (LECO Corporation) for technical advice and support related to instrumentation used in this work and help with data processing.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Polyakova, O.V., Mazur, D.M. & Lebedev, A.T. Improved sample preparation and GC–MS analysis of priority organic pollutants. Environ Chem Lett 12, 419–427 (2014). https://doi.org/10.1007/s10311-014-0464-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10311-014-0464-4