
Abstract
For recombinant production of squalene, which is a triterpenoid compound with increasing industrial applications, in microorganisms generally recognized as safe, we screened Saccharomyces cerevisiae strains to determine their suitability. A strong strain dependence was observed in squalene productivity among Saccharomyces cerevisiae strains upon overexpression of genes important for isoprenoid biosynthesis. In particular, a high level of squalene production (400 ± 45 mg/L) was obtained in shake flasks with the Y2805 strain overexpressing genes encoding a bacterial farnesyl diphosphate synthase (ispA) and a truncated form of hydroxyl-3-methylglutaryl-CoA reductase (tHMG1). Partial inhibition of squalene epoxidase by terbinafine further increased squalene production by up to 1.9-fold (756 ± 36 mg/L). Furthermore, squalene production of 2011 ± 75 or 1026 ± 37 mg/L was obtained from 5-L fed-batch fermentations in the presence or absence of terbinafine supplementation, respectively. These results suggest that the Y2805 strain has potential as a new alternative source of squalene production.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Squalene (2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene; C30H50) is a linear polyunsaturated triterpene with common uses mainly related to the cosmetics industry as a moisturizing agent and emollient. Recently, squalene attracted attention due to multiple studies showing its therapeutic effects and pharmaceutical applications [32, 35], including significant antitumor activities [22, 28, 33]. In addition, it has also been used as a common method of delivering drugs into cells (squalenoylation) [7] and as an emollient in adjuvants for vaccines [9].
The major commercial sources of squalene are liver oil from deep-sea sharks and certain plant-seed oils. However, continuous supply of the liver oils is uncertain due to environmental concerns, and the supply of plant-seed oils is also uncertain due to the low squalene yield and unstable production in certain plant species [24, 36, 39]. Recently, microalgae and other microorganisms have emerged as new alternative sources for squalene production. Among these microorganisms, yeast is a good alternative due to the ease of genetic and physiological manipulation necessary for squalene production. In particular, Saccharomyces cerevisiae is an attractive microorganism for the production of isoprenoids, such as squalene, due to its intrinsically large pools of precursors [20, 21] and its status as generally recognized as safe (GRAS). Moreover, the food-grade status of microorganisms is an essential prerequisite when industrial production is intended for human consumption. Therefore, S. cerevisiae have been successfully employed in metabolic engineering for the production of sterols [42].
In yeast S. cerevisiae, squalene is synthesized as the first precursor via the mevalonate pathway to produce sterols, such as ergosterol (Fig. 1) [6]. 3-Hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase and squalene synthase are the major control enzymes for squalene production in the yeast metabolic pathway [6]. Overexpression of a cytosolic truncated form of HMG-CoA reductase (encoded by tHMG1) leads to squalene accumulation in S. cerevisiae [27]. In a recent study, Mantzouridou and Tsimidou (2010) reported that an extra copy of the HMG2 gene with a K6R-stabilizing mutation induced a strong increase in the size of the squalene pool [21]. In addition, numerous metabolic engineering studies have been performed to improve the production of squalene in S. cerevisiae (summarized in Table 1). Based on these studies, squalene productivity shows a remarkable degree of strain dependence [25], with large variability in squalene production levels appearing to be a consequence of different genetic backgrounds in the strains.

Description of the ergosterol-biosynthesis pathway in Saccharomyces cerevisiae. Abbreviations of the pathway intermediates and genes are as follows: IPP isopentenyl diphosphate, DMAPP dimethylallyl diphosphate, GPP geranyl diphosphate, FPP farnesyl diphosphate, mvaE fused acetoacetyl-CoA thiolase/HMG-CoA reductase gene from Enterococcus faecalis, tHMG1 a truncated form of HMG-CoA reductase, IDI1 IPP isomerase, ERG20 farnesyl diphosphate synthase, ispA farnesyl diphosphate synthase from Escherichia coli, GPPS2 geranyl diphosphate synthase gene from Abies grandis, ERG9 squalene synthase ERG1 squalene epoxidase. Underlined genes represent engineering targets used in this study. Broken arrows represent multiple enzymatic steps and unbroken arrows represent a single enzymatic step. The broken lined box indicates a specific inhibitor
In this study, we screened several S. cerevisiae strains with high potential for squalene accumulation to determine a suitable host for recombinant production of squalene. The selected strain, Y2805 overexpressing tHMG1 and bacterial farnesyl pyrophosphate (FPP) synthase (ispA), produced a 1.7-fold higher yield of squalene as compared with strain BY4741 which has been most frequently employed as the host for metabolic engineering related to squalene overproduction. The suitability of Y2805 as a platform strain for large-scale production was confirmed through fed-batch fermentation experiments under optimal conditions. During the course of this study, we noted that commonly used protocols for the determination of squalene content in S. cerevisiae showed variable results. Therefore, we compared the different protocols for squalene determination frequently used in the literature and proposed simple and efficient methods for squalene extraction from S. cerevisiae cells.
Materials and methods
Strains and plasmids
Strains and plasmids used in this study are listed in Table 2. The expression plasmid pYEGα and the host strain S. cerevisiae Y2805 were described previously [2, 34]. S. cerevisiae ATCC200589, ATCC201238, ATCC201741, BY4741, and W303 were obtained from the American Type Culture Collection (Manassas, VA, USA), and S. cerevisiae YPH499 was purchased from Stratagene (La Jolla, CA, USA). Escherichia coli DH5α was used for plasmid construction and propagation.
Plasmid construction
The primers designed for plasmid construction are listed in Table 3. The reagents for molecular manipulations were obtained from Takara (Shiga, Japan) and Qiagen (Valencia, CA, USA). DNA fragments were amplified by polymerase chain reaction (PCR) using a Phusion Hot Start II high-fidelity DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The tHMG1 (a truncated form of HMG-CoA reductase lacking the regulatory membrane-spanning domain), yeast farnesyl diphosphate synthase (ERG20), and squalene synthase (ERG9) genes were amplified using corresponding primers, digested with EcoRI and SalI (XhoI for ERG20), and placed downstream of the GAL10 promoter by cloning into the EcoRI and SalI sites of pYEGα to obtain plasmids pG-tHMG1, pG-ERG20, and pG-ERG9, respectively. The DNA sequences of acetyl-CoA acetyltransferase/HMG-CoA reductase (mvaE) from Enterococcus faecalis, ispA from E. coli, and geranyl diphosphate synthase (GPPS2) from Abies grandis were codon optimized for expression in yeast and synthesized (GenScript Corp., Piscataway, NJ, USA). The synthesized mvaE, ispA, and GPPS2 genes were digested with EcoRI/SalI and ligated into the corresponding sites in pYEGα to obtain plasmids pG-mvaE, pG-ispA, and pG-GPPS2, respectively. To construct plasmids pG-H-I and pG-H-E20, cloning was performed using the InFusion HD cloning kit (Takara). First, the tHMG1 fragment, including promoter (521 bp) and terminator (427 bp) regions, was amplified from plasmid pG-tHMG1 using the primer pair Infusion smaI-F and Infusion smaI-R. The PCR product was inserted into the SmaI-digested pG-ispA and pG-ERG20 plasmids by homologous recombination, generating plasmids pG-H-I and pG-H-E20, respectively. Combinations of other genes were constructed using the same method.
Chemicals, media, and culture conditions
All chemical reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA) and BD Bioscience (Franklin Lakes, NJ, USA). UD medium (20-g/L glucose, 6.7-g/L yeast nitrogen base without amino acids, and 0.77-g/L Ura dropout supplement mixture) was used for transformant selection and pre-cultures of recombinant yeasts harboring episomal plasmids. YPD medium (10-g/L yeast extract, 20-g/L peptone, and 20-g/L glucose) and YPDG medium (10-g/L yeast extract, 20-g/L peptone, 10-g/L glucose, and 10-g/L galactose) were used for fermentation experiments in baffled flasks, with galactose used for induction of genes under the control of galactose-inducible promoters. The plasmids were transformed into the S. cerevisiae strains using the LiAc/TE method [13], and cells were plated on UD agar medium for selection of transformants. The colonies formed on the plates were checked by PCR for the presence of the correct plasmids. Test-tube cultures containing 3 mL of UD medium were used as seed cultures and were grown overnight at 30 °C before use to inoculate cultures in 250-mL baffled flasks containing 25 mL of medium at an OD600 of 0.1. Flask cultures were performed in triplicate at 30 °C and 200 rpm in a shaking incubator (HB-201MS-2R; Hanbaek Scientific Co., Bucheon, Korea) for 72 h.
Fed-batch fermentation
Fed-batch fermentations were performed in a 5-L fermenter (CNS, Daejeon, Korea), with an initial medium comprising 10-g/L yeast extract, 20-g/L peptone, and 20-g/L glucose. A pre-culture was prepared by inoculating a single colony into 25 mL of UD medium for 24 h, followed by transfer of the culture into a 1-L baffled flask containing 200 mL of YPD medium and growth for 16 h. The seed culture was then inoculated into a 5-L fermenter containing 2-L medium. Fed-batch fermentations were performed at 30 °C with aeration at 1 vvm and agitation at 600 rpm was maintained using two six-bladed impellers. Culture pH was maintained at 6.0 by automatic feeding of a 10-N NaOH solution. Dissolved oxygen was monitored using OxyProbe (Broadley-James, Irvine, CA, USA) and maintained at > 30% saturation with an agitation cascade. The temperature, air-flow rate, agitation, pH, and dissolved oxygen were monitored and controlled using CNS monitoring and control systems (CNS). A feeding media comprising 600-g/L glucose and 40-g/L yeast extract was fed to the 5-L fermenter at specific rates of between 0-g/L/h and 5-g/L/h following depletion of the initially added glucose. Galactose was added as an inducer every 24 h at 5 g/L, and antifoam 204 (Sigma-Aldrich) was added manually when foam was generated during fermentation. The culture was sampled every 12 h to measure cell growth, glucose concentration, and squalene production.
Squalene analysis
Four different protocols for squalene extraction in S. cerevisiae were tested: the hot alcoholic KOH–dodecane method [30], the hot HCl–acetone method [10], the bead-methanolic KOH–hexane method [41], and the bead–pentane method (this study). The procedure for the hot alcoholic KOH-dodecane method is as follows. Cells from 2 mL of the yeast culture were collected by centrifugation at 15,000g and resuspended in 0.4 mL of the alcoholic KOH solution. Resuspended cells were placed into boiling water for 5 min and allowed to cool on ice. Dodecane (0.4 mL) was added to the resuspended cells and vortexed vigorously for 5 min. After centrifugation at 3,000g for 5 min, the dodecane layer was analyzed by gas chromatography (GC). For the hot HCl-acetone method, cells were pelleted and treated with 3-M HCl at 95 °C for 5 min, chilled on ice for 10 min, and then washed with distilled water before extracting squalene with 1 mL of acetone. The extracts were centrifuged at 13,000g for 5 min, and the supernatants were analyzed by high-performance liquid chromatography (HPLC) using an Agilent 1200 instrument equipped with a C18 column and a UV detector (Agilent Technologies, Santa Clara, CA, USA). For the bead-methanolic KOH-hexane method, cells were harvested by centrifugation for 5 min, washed with deionized water, and disrupted with glass beads. The cell homogenate was incubated in three volumes of methanolic KOH at 70 °C for 2 h. The saponification mixture was extracted twice with three volumes of hexane, and the combined extracts were dried under nitrogen. The lipid residue containing squalene was dissolved in a desired volume of hexane, and the sample was analyzed by HPLC. The detailed procedures of these protocols were described previously [10, 30, 41] and the bead–pentane method used in this study is subsequently described here in detail.
For the bead–pentane protocol, cells from 1 mL of yeast culture were pelleted and resuspended in 600 µL of 50-mM Tris–HCl (pH 7.5). Glass beads (425–600 μm; Sigma-Aldrich) were added to the resuspended cells, disrupted by vortexing vigorously for 10 min, and interspersed with cooling on ice for 5 min. Pentane (400 µL) containing 0.1% (v/v) methyl laurate as an internal standard was added to the resuspended cells, mixed by vortexing for 10 min, and centrifuged at 10,000g for 10 min. When necessary, a saponification step (boiling with 0.4 mL of alcoholic KOH) described in the first protocol was included following the bead-beating step. The top pentane layer was analyzed by GC using an Agilent 7890A (Agilent Technologies) equipped with a flame-ionization detector and an HP-5 capillary column (30 m × 0.25 μm; Agilent Technologies). The sample (2 µL) from the pentane layer was analyzed in a capillary column with a split ratio of 10 using helium as a carrier gas and at a flow rate of 1 mL/min. The GC oven temperature was initially held at 40 °C for 3 min, followed by an increase to 240 °C at a rate of 10 °C/min where it was maintained for 5 min until increasing to 300 °C at a rate of 10 °C/min and maintained for 1 min. The injector temperature was 300 °C and the detector temperature was 320 °C. Squalene quantification was performed using standard curves. All analyses were performed in duplicate.
Results and discussion
Basal levels of squalene in non-engineered S. cerevisiae strains
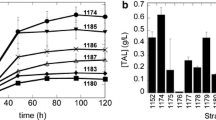
In S. cerevisiae, squalene biosynthesis is tightly regulated and accumulates to a negligible level during normal growth. Under normal conditions, yeast cells rapidly metabolize squalene to sterols, such as ergosterol, which is an essential component for maintaining yeast-cell viability and is synthesized from squalene as the first precursor. Many enzymes are involved in the squalene conversion to ergosterol, but as the first step of the ergosterol–biosynthesis pathway, squalene epoxidase encoded by ERG1 is the key enzyme necessary for increased squalene production. Partial inhibition of squalene epoxidase activity with the addition of its inhibitor, terbinafine, resulted in squalene accumulation [8, 11, 23]. The basal levels of squalene reported for different strains in the absence or presence of terbinafine are summarized in Table 4 and ranged from 0.056- to 0.8-mg/g dry cell weight (DCW) and from 0.5- to 34-mg/L in the absence of terbinafine. In the presence of terbinafine, squalene accumulation increased from 0.31 to 10.02 mg/g DCW or from 34 to 119.8 mg/L. In this study, we compared strains of S cerevisiae (BY4741, W303, and YPH499) frequently used for squalene accumulation (Tables 1 and 4) and ATCC strains (ATCC200589, 201238, and 201741) reportedly exhibiting comparatively enhanced levels of squalene accumulation among the 37 strains tested upon HMG1 overexpression (Table 1) [25]. Our laboratory strain, Y2805, which has been previously used for isoprenoid production (unpublished data), was also included for comparison. The level of squalene accumulation among the seven strains in the presence or absence of terbinafine was determined in shake-flask experiments. The squalene content of the seven strains ranged from 0.3 ± 0.01 to 1.4 ± 0.3 mg/L in the absence of terbinafine and from 11.4 ± 0.3 (BY4741) to 40.5 ± 4.5 mg/L (W303) in the presence of terbinafine when assayed using the bead–pentane method (a comparison of the squalene-determination protocols is provided later) (Fig. 2). We noted that despite the more frequent use of BY4741 and 4742 as hosts for metabolic engineering to improve squalene production (Table 1), the non-engineered BY4741 strain showed the lowest level of squalene accumulation, even in the presence of terbinafine (Fig. 2). By contrast, the W303 strain showed the highest level of squalene accumulation in the presence of terbinafine; however, following metabolic engineering, the level of squalene accumulation was negligible (more details are provided later). These contradictory results suggest that squalene accumulation in yeast was highly dependent upon strains with different genetic backgrounds, and that other yeast strains might also represent potential as hosts for squalene production.

Squalene accumulation in non-engineered Saccharomyces cerevisiae strains in the absence and presence of terbinafine. S. cerevisiae strains were grown for 72 h in YPD medium containing terbinafine in shake flasks. White bars indicate squalene production by S. cerevisiae strains grown in medium without terbinafine (control). Black bars indicate squalene production by S. cerevisiae strains grown in medium containing 10 μg/mL terbinafine. Error bars represent the standard deviation from three independent cultivations
Overexpression of genes involved in the early steps of ergosterol biosynthesis in S. cerevisiae
Although S. cerevisiae is an attractive host for the production of isoprenoids, such as squalene, due to the intrinsically large pools of isoprenoid precursors, these intermediates do not usually accumulate in the cell. To overproduce squalene, we overexpressed enzymes required for the biosynthesis of important intermediates (mevalonate and prenyl diphosphate) during the early steps of isoprenoid biosynthesis. Therefore, the genes mvaE, tHMG1, ERG20, ispA, and GPPS2 were overexpressed in S. cerevisiae Y2805 and evaluated as potential key enzyme for squalene production (Fig. 1). In addition, ERG9, which is necessary for squalene synthesis in S. cerevisiae, was also tested. The genes encoding these enzymes were cloned in a yeast-expression plasmid (pYEGα) in various combinations, and S. cerevisiae Y2805 strains harboring these plasmids were cultivated in a shake flask containing YPDG medium for 72 h. As shown in Fig. 3a, S. cerevisiae Y2805 strains overexpressing ERG20, mvaE, ispA, or GPPS2 alone did not produce any detectable amount of squalene, whereas Y2805 strain overexpressing tHMG1, the gene involved in the first committed step of ergosterol biosynthesis, produced high levels of squalene (200 ± 17 mg/L). The effect of tHMG1 overexpression on squalene accumulation was also tested in other commonly used S. cerevisiae strains, including W303 and YPH499. In contrast to the high degree of squalene accumulation in Y2805, levels in W303 and YPH499 were negligible (2.4 ± 0.4 and 9.2 ± 1.5 mg/L, respectively). Numerous reports previously showed that tHMG1 overexpression results in improved squalene production in yeast (Table 1). Moreover, compared with the wild-type EGY48 strain, squalene production increased by 20-fold in tHMG1-overexpressing variants [21]. Polakowski et al. (1998) also reported that squalene accumulation was increased by overexpressing the truncated HMG1 gene [27].

Comparison of squalene production in Saccharomyces cerevisiae Y2805 overexpressing various genes. Squalene production by S. cerevisiae Y2805 overexpressing individual genes (a) and various combinations (b) after a 72-h cultivation. Strains were grown at 30 °C and 200 rpm in YPDG medium for 72 h. Abbreviations of plasmids are as follows: pG-M-I overexpressing mvaE and ispA, pG-M-G overexpressing mvaE and GPPS2, pG-H-I overexpressing tHMG1 and ispA; pG-H-E20 overexpressing tHMG1 and ERG20, pG-H-G overexpressing tHMG1 and GPPS2, pG-H-E9 overexpressing tHMG1 and ERG9. Error bars represent the standard deviation from three independent cultivations
Co-expression of the ispA or ERG20 gene with the tHMG1 gene further increased squalene production (Fig. 3b). In particular, the Y2805 strain harboring pG-H-I (co-overexpression of the tHMG1 and ispA genes) resulted in a significant increase in squalene production by up to twofold (400 ± 45 mg/L) as compared with the same strain overexpressing tHMG1 alone. Wang et al. (2010) also reported that overexpression of the ispA gene significantly increased farnesol production in an E. coli incorporating the exogenous mevalonate pathway. However, overexpression of GPPS2 or ERG9 along with tHMG1 was ineffective for squalene accumulation, resulting in squalene production levels lower than those observed in strains overexpressing tHMG1 alone. These results suggested that truncated HMG-CoA reductase encoded by tHMG1 and FPP synthase encoded by ispA were the two key enzymes involved in the isoprenoid-biosynthesis pathway for production of isoprenoids, such as squalene.
Comparative evaluation of protocols for squalene determination in S. cerevisiae
Several protocols for squalene determination in S. cerevisiae are available in the literature [10, 30, 41], and comprise steps for cell breakage, saponification, and solvent extraction, although, in some cases, the saponification step is omitted. Cell breakage is commonly performed by bead-beating with glass beads, whereas some protocols describe boiling in the presence of strong acid or alkali to omit the bead-beating step. Here, we compared protocols commonly used for squalene determination in S. cerevisiae and also compared these with our new protocol. Four different protocols were evaluated using S. cerevisiae strain Y2805 overexpressing tHMG1 and ispA. Two protocols, the hot alcoholic KOH-dodecane [30] and hot HCl-acetone [10] methods, omit the bead-beating step for cell breakage and instead employ treatment with boiling alcoholic KOH or hot HCl, followed by solvent extraction with dodecane or acetone. The boiling alcoholic KOH treatment in the former protocol appears to serve also as the saponification step. The other two protocols include bead-beating with glass beads for cell breakage in the presence or absence of the saponification step. The third protocol (bead-methanolic KOH-hexane) [41] consists of separate steps of cell disruption with bead-beating, saponification with hot methanolic KOH, and extraction with hexane. The final protocol (bead–pentane) developed in this study included cell breakage with bead-beating and extraction with pentane.
As shown in Table 5, the hot alcoholic KOH-dodecane protocol yielded considerably lower efficiency of squalene extraction (27.7 ± 2.8 mg/L) as compared with other protocols, likely due to the omission of physical disruption of the cell. By contrast, the hot HCl-acetone protocol resulted in a good extraction efficiency (317 ± 27 mg/L), but also not as good as that observed from protocols involving bead-beating. The addition of a cell breakage step (bead-beating with glass beads) replacing the boiling treatment in the hot alcoholic KOH-dodecane protocol greatly improved squalene extraction, resulting in levels (384 ± 13 mg/L) comparable with those obtained in other protocols. Therefore, these findings suggested that the bead-beating process frequently used for cell breakage in yeast-cell biology is a prerequisite for efficient extraction of squalene from yeast cells, and that this process cannot be adequately replaced with the use of strong acid or alkali treatment with heating. The bead–pentane protocol appeared to be the most efficient method, yielding 400 ± 35 mg/L. Notably, the omission of the saponification step did not influence the efficiency of squalene extraction (395 ± 10 vs. 400 ± 35 mg/L); however, the choice of extraction solvent did influence efficiency, with pentane being slightly superior to dodecane (404 ± 41 vs. 384 ± 13 mg/L and 400 ± 35 vs. 379 ± 24 mg/L).
These data suggested that the bead–pentane protocol proposed in this study might serve as a simple and efficient method for squalene extraction from S. cerevisiae cells. Spiking with a known amount of squalene in the bead–pentane protocol showed an excellent recovery percentage (97%), which confirmed its potential use as a simple and efficient method for squalene extraction from yeast. Spiking experiments in the other protocols suggested that the additional squalene appeared to be lost (20–30%) during this process, possibly explaining the poor extraction yields from these two methods (hot alcoholic KOH-dodecane and bead-methanolic KOH-hexane).
Strain dependence for squalene accumulation from engineered S. cerevisiae strains overexpressing tHMG1 and ispA
Using the new protocol described here, the strains employed for testing basal squalene levels were evaluated for their potential use as hosts for squalene accumulation following metabolic engineering. The ATCC strains 200589, 201238, and 201741, which reportedly accumulated high levels of squalene when engineered to overexpress HMG1 [25], were selected for this test, and the BY4741 strain, the most frequently used host for engineering the squalene-biosynthesis pathway, was also included. The YPH499 and W303 strains were not tested, because squalene accumulation was negligible in these strains following engineering for tHMG1 overexpression.
The ATCC strains and BY4741 were transformed with the plasmid pG-H-I, which resulted in the highest squalene production, and their squalene production levels were determined after cultivation in a shake flask for 72 h and compared with those from the Y2805 strain containing the same plasmid. As shown in Fig. 4a, recombinant strains harboring pG-H-I exhibited higher squalene production as compared with wild-type strains without recombinant plasmids. The Y2805 strain produced the highest squalene levels (400 ± 35 mg/L), followed by BY4741, which produced 240 ± 15 mg/L squalene. The three ATCC strains (ATCC200589, ATCC201238, and ATCC201741) harboring pG-H-I produced a considerably lower amounts of squalene (5.2 ± 1.6, 1.6 ± 0.5, and 12.8 ± 2.0 mg/L, respectively) as compared with the other two strains (BY4741 and Y2805) harboring the same plasmid. Despite the non-engineered BY4741 strain showing the lowest squalene accumulation in the presence of terbinafine, the metabolically engineered BY4741 strain showed a considerably higher squalene level as compared with the ATCC strains. Therefore, determining the level of squalene accumulation in non-engineered strains in the presence of terbinafine did not represent a criterion for the selection of host strains for engineering yeast metabolic pathways for squalene biosynthesis.

Optimization of squalene production in Saccharomyces cerevisiae strains. a Comparison of squalene production by different S. cerevisiae strains. White bars indicate squalene production in wild-type yeast strains, and black bars indicate squalene production in yeast recombinant strains harboring pG-H-I. b Effect of cultivation temperature on squalene production in S. cerevisiae Y2805 harboring pG-H-I. c Inhibition effect of terbinafine in S. cerevisiae harboring pG-H-I. White bars indicate cell growth of Y2805 and black bars indicate squalene production in Y2805. Strains were grown in YPDG medium for 72 h in all cases. Error bars represent the standard deviation from three independent cultivations
Our results showed that Y2805 was the optimal strain among the seven yeast strains tested for squalene production. To investigate variations in genetic background possibly responsible for this pronounced difference in squalene accumulation among strains, the nucleotide sequences of the relevant genes important for squalene biosynthesis were determined for Y2805 and ATCC200589. The genes (ERG9, ERG1, and MGA2) did not show any variations in nucleotide sequences in the coding and promoter region between each other or as compared with the standard strain (288C). Therefore, it remains unclear why Y2805 accumulated considerably higher amounts of squalene as compared with the other tested strains. Further studies at the levels of transcription and translation along with other characterizations are required.
Optimization of fermentation conditions for squalene production
Prior to fermentation studies, fermentation conditions, especially with respect to the temperature, were optimized in flask experiments. According to the previous studies, squalene levels in yeast cells are altered by changing culture conditions, such as media composition and cultivation temperature [12, 19, 38]. Among these, temperature optimization is a facile method used to improve the production of desired compounds and affect cell growth. Recently, Henderson et al. (2013) reported that ethanol fermentation using S. cerevisiae strains (Prise de Mousse and Sake A18) increased squalene production by up to twofold at 35 °C [12], whereas Loertscher et al. (2006) and Tronchoni et al. (2012) reported that cultivation of S. cerevisiae (JRY527 and T73, respectively) at low temperatures accumulated high levels of squalene [19, 38]. Therefore, we tested the effect of high (35 °C) and low (25 °C) temperatures on squalene production and cell growth in Y2805 strains harboring pG-H-I. However, in contrast to the previous reports, cultivation of Y2805 overexpressing the tHMG1 and ispA genes resulted in significantly decreased squalene levels (156 ± 19 mg/L) at 25 °C and also reduced squalene accumulation (277 ± 47 mg/L) at 35 °C as compared with cultivation at 30 °C as a control (Fig. 4b). Similar to these results, S. cerevisiae KLN1-ERG1 harboring a chromosomal copy of the ERG1 gene disrupted and transformed with a plasmid containing ERG1, also showed significantly reduced squalene levels at low (22 °C) and high (35 °C) temperatures [11]. These results indicated that optimization of culture conditions, such as cultivation temperature, for squalene production in S. cerevisiae appeared specific to strains harboring different genetic backgrounds.
As described in a previous section, partial inhibition of squalene epoxidase by terbinafine in non-engineered S. cerevisiae strains, including Y2805, results in a considerable increase in squalene accumulation within cells. Based on this result, we tested the possibility for further improvements in squalene productivity by inhibiting squalene epoxidase with terbinafine in the engineered Y2805 strain (transformed with pG-H-I). Y2805 harboring pG-H-I was cultivated in YPDG medium containing different concentrations of terbinafine (5–40 μg/mL) for 72 h. As shown in Fig. 4c, squalene levels in cells gradually increased along with the concentration of terbinafine. In particular, when terbinafine was added at a concentration of 20 μg/mL, the highest level of squalene (804 ± 31 mg/L) was obtained, although the growth of terbinafine-treated cells was slightly effected. However, treatment with 10 μg/mL of terbinafine did not affect the growth of yeast cells and showed a similar level of squalene accumulation (756 ± 36 mg/L). Increasing the concentration of terbinafine to 40 μg/mL was expected to result in a higher squalene accumulation; however, the growth of terbinafine-treated cells was significantly inhibited, and squalene levels were, therefore, considerably lower (531 ± 24 mg/L). The terbinafine concentration (10 μg/mL) that did not affect cell growth was selected as the optimal concentration and subsequently used for squalene production during fermentation. Terbinafine treatment during fermentation with Y2805 harboring pG-H-I increased squalene accumulation by up to twofold, indicating that squalene epoxide activity in Y2805 dissipated half the amount of accumulated squalene further downstream to ergosterol. These results also suggested that strain development to contain diminished squalene epoxidase activity might partially replace the need for terbinafine treatment.
Squalene production in 5-L fed-batch fermentation
Based on the results obtained from flask cultivation under various conditions, we chose the best squalene-producing strain (Y2805 transformed with pG-H-I) and performed fed-batch fermentation in a 5-L fermenter with a working volume of 2 L. To evaluate the performance of S. cerevisiae Y2805 transformed with pG-H-I for squalene production, 20 g/L of glucose was used as the initial carbon source, and glucose feeding was initiated after the initial glucose was consumed. Galactose was also added at regular intervals every 24 h to induce genes under the control of a galactose-inducible promoter. The glucose-feeding rate was specifically controlled to prevent carbon catabolite repression caused by the presence of high concentrations of glucose in the medium. As shown in Fig. 5, squalene production began within 24 h and gradually increased as along with cell growth. Consequently, during 7 days of fermentation, we obtained a maximum squalene production of 1026 ± 37 mg/L at a cell density of OD600 = 152 at 144 h (Fig. 5a). This production level corresponded to a 2.5-fold increase relative to the highest level obtained from shake-flask cultivation. Similar to fed-batch fermentation performed to evaluate squalene production in the absence of terbinafine, fed-batch fermentation was conducted to measure squalene accumulation using the same recombinant strain and in the presence of 10 μg/mL terbinafine. As shown in Fig. 5b, squalene production was initiated after galactose induction and continued steadily, reaching the highest level of 2011 ± 75 mg/L after 144 h of fermentation, with no significant inhibition of the growth of terbinafine-treated yeast cells. Compared with levels obtained from fermentation of the same recombinant strain in the absence of terbinafine, squalene production increased by ~ twofold. Treatment with 10-μg/mL terbinafine was sufficient to reduce of the flux of accumulated squalene to post-squalene in the ergosterol-biosynthesis pathway, thereby allowing a maximal increase in squalene production (~ twofold higher) relative to that observed in shake-flask experiments. This verified that squalene epoxidase was an important target for improving squalene productivity. Strain improvement involving further diminishing of squalene epoxidase activity by genetic engineering is under way to further improve the development of Y2805 as a suitable host for large-scale squalene production.

Squalene production in fed-batch fermentation using Saccharomyces cerevisiae Y2805 harboring pG-H-I. a Time profile of fed-batch fermentation of S. cerevisiae Y2805 transformed with pG-H-I. b Time profile of fed-batch fermentation of S. cerevisiae Y2805 transformed with pG-H-I in medium containing 10 μg/mL terbinafine. The strain was grown under glucose-limited conditions and galactose was added at regular intervals every 24 h. Typical profiles observed for cell growth (OD600, black circles), glucose concentration (g/L, white circles), feeding rate (g/L/h, lines), and squalene concentration (mg/L, gray bars) are represented. Data were obtained during fed-batch fermentation and represent the average values from two independent analyses
In summary, this study reported considerable strain dependence in squalene accumulation among different strains of S. cerevisiae, and the metabolically engineered S. cerevisiae Y2805 was selected as a new alternative source exhibiting improved squalene production. This study also provided a simple and efficient protocol for the extraction of squalene from S. cerevisiae cells.
References
Asadollahi MA, Maury J, Schalk M, Clark A, Nielsen J (2010) Enhancement of farnesyl diphosphate pool as direct precursor of sesquiterpenes through metabolic engineering of the mevalonate pathway in Saccharomyces cerevisiae. Biotechnol Bioeng 106:86–96
Choi E-S, Sohn J-H, Rhee S-K (1994) Optimization of the expression system using galactose-inducible promoter for the production of anticoagulant hirudin in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 42:587–594
Dai Z, Liu Y, Huang L, Zhang X (2012) Production of miltiradiene by metabolically engineered Saccharomyces cerevisiae. Biotechnol Bioeng 109:2845–2853
Dai Z, Liu Y, Zhang X, Shi M, Wang B, Wang D, Huang L, Zhang X (2013) Metabolic engineering of Saccharomyces cerevisiae for production of ginsenosides. Metab Eng 20:146–156
Dai Z, Wang B, Liu Y, Shi M, Wang D, Zhang X, Liu T, Huang L, Zhang X (2014) Producing aglycons of ginsenosides in bakers’ yeast. Sci Rep 4:3698
Daum G, Lees ND, Bard M, Dickson R (1998) Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast 14:1471–1510
Desmaële D, Gref R, Couvreur P (2012) Squalenoylation: a generic platform for nanoparticular drug delivery. J Control Release 161:609–618
Drozdíková E, Garaiová M, Csáky Z, Obernauerová M, Hapala I (2015) Production of squalene by lactose-fermenting yeast Kluyveromyces lactis with reduced squalene epoxidase activity. Lett Appl Microbiol 61:77–84
Fox CB (2009) Squalene emulsions for parenteral vaccine and drug delivery. Molecules 14:3286–3312
Gao S, Tong Y, Zhu L, Ge M, Zhang Y, Chen D, Jiang Y, Yang S (2017) Iterative integration of multiple-copy pathway genes in Yarrowia lipolytica for heterologous β-carotene production. Metab Eng 41:192–201
Garaiová M, Zambojová V, Šimová Z, Griač P, Hapala I (2014) Squalene epoxidase as a target for manipulation of squalene levels in the yeast Saccharomyces cerevisiae. FEMS Yeast Res 14:310–323
Henderson CM, Zeno WF, Lerno LA, Longo ML, Block DE (2013) Fermentation temperature modulates phosphatidylethanolamine and phosphatidylinositol levels in the cell membrane of Saccharomyces cerevisiae. Appl Environ Microbiol 79:5345–5356
Holm C, Meeks-Wagner DW, Fangman WL, Botstein D (1986) A rapid, efficient method for isolating DNA from yeast. Gene 42:169–173
Hull CM, Loveridge EJ, Rolley NJ, Donnison IS, Kelly SL, Kelly DE (2014) Co-production of ethanol and squalene using a Saccharomyces cerevisiae ERG1 (squalene epoxidase) mutant and agro-industrial feedstock. Biotechnol Biofuels 7:133
Kirby J, Romanini DW, Paradise EM, Keasling JD (2008) Engineering triterpene production in Saccharomyces cerevisiae–β-amyrin synthase from Artemisia annua. FEBS J 275:1852–1859
Kuranda K, Grabinska K, Berges T, Karst F, Leberre V, Sokol S, François J, Palamarczyk G (2009) The YTA7 gene is involved in the regulation of the isoprenoid pathway in the yeast Saccharomyces cerevisiae. FEMS Yeast Res 9:381–390
Kwak S, Kim SR, Xu H, Zhang GC, Lane S, Kim H, Jin YS (2017) Enhanced isoprenoid production from xylose by engineered Saccharomyces cerevisiae. Biotechnol Bioeng 114:2581–2591
Liu J, Zhang W, Du G, Chen J, Zhou J (2013) Overproduction of geraniol by enhanced precursor supply in Saccharomyces cerevisiae. J Biotechnol 168:446–451
Loertscher J, Larson LL, Matson CK, Parrish ML, Felthauser A, Sturm A, Tachibana C, Bard M, Wright R (2006) Endoplasmic reticulum-associated degradation is required for cold adaptation and regulation of sterol biosynthesis in the yeast Saccharomyces cerevisiae. Eukaryot Cell 5:712–722
Mantzouridou F, Naziri E, Tsimidou MZ (2009) Squalene versus ergosterol formation using Saccharomyces cerevisiae: combined effect of oxygen supply, inoculum size, and fermentation time on yield and selectivity of the bioprocess. J Agric Food Chem 57:6189–6198
Mantzouridou F, Tsimidou MZ (2010) Observations on squalene accumulation in Saccharomyces cerevisiae due to the manipulation of HMG2 and ERG6. FEMS Yeast Res 10:699–707
Murakoshi M, Nishino H, Tokuda H, Iwashima A, Okuzumi J, Kitano H, Iwasaki R (1992) Inhibition by squalene of the tumor-promoting activity of 12-O-tetradecanoylphorbol-13-acetate in mouse-skin carcinogenesis. Int J Cancer 52:950–952
Naziri E, Mantzouridou F, Tsimidou MZ (2011) Enhanced squalene production by wild-type Saccharomyces cerevisiae strains using safe chemical means. J Agric Food Chem 59:9980–9989
Newmark HL (1997) Squalene, olive oil, and cancer risk: a review and hypothesis. Cancer Epidemiol Biomarkers Prev 6:1101–1103
Ohto C, Muramatsu M, Obata S, Sakuradani E, Shimizu S (2009) Overexpression of the gene encoding HMG-CoA reductase in Saccharomyces cerevisiae for production of prenyl alcohols. Appl Microbiol Biotechnol 82:837–845
Paradise EM, Kirby J, Chan R, Keasling JD (2008) Redirection of flux through the FPP branch-point in Saccharomyces cerevisiae by down-regulating squalene synthase. Biotechnol Bioeng 100:371–378
Polakowski T, Stahl U, Lang C (1998) Overexpression of a cytosolic hydroxymethylglutaryl-CoA reductase leads to squalene accumulation in yeast. Appl Microbiol Biotechnol 49:66–71
Rao CV, Newmark HL, Reddy BS (1998) Chemopreventive effect of squalene on colon cancer. Carcinogenesis 19:287–290
Rasool A, Ahmed MS, Li C (2016) Overproduction of squalene synergistically downregulates ethanol production in Saccharomyces cerevisiae. Chem Eng Sci 152:370–380
Rodriguez S, Kirby J, Denby CM, Keasling JD (2014) Production and quantification of sesquiterpenes in Saccharomyces cerevisiae, including extraction, detection and quantification of terpene products and key related metabolites. Nat Protoc 9:1980–1996
Sere YY, Regnacq M, Colas J, Berges T (2010) A Saccharomyces cerevisiae strain unable to store neutral lipids is tolerant to oxidative stress induced by α-synuclein. Free Radic Biol Med 49:1755–1764
Smith TJ (2000) Squalene: potential chemopreventive agent. Expert Opin Investig Drugs 9:1841–1848
Smith TJ, Yang G, Seril DN, Liao J, Kim S (1998) Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis by dietary olive oil and squalene. Carcinogenesis 19:703–706
Sohn J-H, Lee S-K, Choi E-S, Rhee S-K (1991) Gene expression and secretion of the anticoagulant hirudin in Saccharomyces cerevisiae. J Microbiol Biotechnol 1:266–273
Spanova M, Daum G (2011) Squalene–biochemistry, molecular biology, process biotechnology, and applications. Eur J Lipid Sci Technol 113:1299–1320
Storelli M, Ceci E, Storelli A, Marcotrigiano G (2003) Polychlorinated biphenyl, heavy metal and methylmercury residues in hammerhead sharks: contaminant status and assessment. Mar Pollut Bull 46:1035–1039
Tokuhiro K, Muramatsu M, Ohto C, Kawaguchi T, Obata S, Muramoto N, Hirai M, Takahashi H, Kondo A, Sakuradani E (2009) Overproduction of geranylgeraniol by metabolically engineered Saccharomyces cerevisiae. Appl Environ Microbiol 75:5536–5543
Tronchoni J, Rozès N, Querol A, Guillamón JM (2012) Lipid composition of wine strains of Saccharomyces kudriavzevii and Saccharomyces cerevisiae grown at low temperature. Int J Food Microbiol 155:191–198
Turoczy N, Laurenson L, Allinson G, Nishikawa M, Lambert D, Smith C, Cottier J, Irvine S, Stagnitti F (2000) Observations on metal concentrations in three species of shark (Deania calcea, Centroscymnus crepidater, and Centroscymnus owstoni) from southeastern Australian waters. J Agric Food Chem 48:4357–4364
Valachovic M, Garaiova M, Holic R, Hapala I (2016) Squalene is lipotoxic to yeast cells defective in lipid droplet biogenesis. Biochem Biophys Res Commun 469:1123–1128
Valachovič M, Hapala I (2017) Biosynthetic approaches to squalene production: the case of yeast. Methods Mol Biol 1494:95–106
Veen M, Stahl U, Lang C (2003) Combined overexpression of genes of the ergosterol biosynthetic pathway leads to accumulation of sterols in Saccharomyces cerevisiae. FEMS Yeast Res 4:87–95
Xie W, Lv X, Ye L, Zhou P, Yu H (2015) Construction of lycopene-overproducing Saccharomyces cerevisiae by combining directed evolution and metabolic engineering. Metab Eng 30:69–78
Zhuang X, Chappell J (2015) Building terpene production platforms in yeast. Biotechnol Bioeng 112:1854–1864
Acknowledgements
This research was supported by a Grant from the KRIBB Research Initiative Program and by a National Research Foundation of Korea (NRF) Grant from the Korea government (MSIP) (Grant no. NRF-2016R1A2B4009432). This work was also supported by the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry, and Fisheries (IPET) through the Animal Disease Management Technology Development Program funded by the Ministry of Agriculture, Food, and Rural Affairs (MAFRA; 316043-3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Han, J.Y., Seo, S.H., Song, J.M. et al. High-level recombinant production of squalene using selected Saccharomyces cerevisiae strains. J Ind Microbiol Biotechnol 45, 239–251 (2018). https://doi.org/10.1007/s10295-018-2018-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-018-2018-4