
Abstract
The gene integration method is an important tool to stably express desirable genes in bacteria. To avoid heavy workload and cost, we constructed a rapid and efficient method for genome modification. This method depended on a mobilizable plasmid, which contains a P tac promoter, an introduced multiple cloning site (iMCS), and rrnBT1T2 terminator. Briefly, the mobilizable plasmid pK18-MBPMT with the P tac-iMCS-rrnBT1T2 cartridge derived from pK18mobsacB was prepared to directly integrate hetero-/homologous DNA into the Corynebacterium glutamicum genome. Like our previous method, this method was based on insertional inactivation and double-crossover homologous recombination, which simultaneously achieved gene overexpression and inactivation in the genome without the use of genetic markers. Compared to the previous method, this protocol omitted the construction of a recombinant expression plasmid and clone of the target gene(s) cassette, which significantly decreased the workload, cost, and operational time. Using this method, the heterologous gene amy and the homologous gene lysC T311I were successfully integrated into the C. glutamicum genome at alaT and avtA loci, respectively. Moreover, the operation time of this method was shorter than that of the previous method, especially for repeated integration. This method, which is based on the mobilizable plasmid pK18-MBPMT, thus represents a potentially attractive protocol for the integration of genes in the course of genetic modification of C. glutamicum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Regulation of gene expression in Corynebacterium glutamicum is one of the hot research areas, because this non-pathogenic and Gram-positive bacterium has shown to be a versatile microorganism. C. glutamicum has been widely used in large-scale fermentative production of many biochemicals, such as amino acids, nucleic acids, and organic acids [1, 2]. Metabolic engineering via genetic modification in C. glutamicum is based on intensifying or shifting carbon flux towards the biosynthetic pathway of the aimed product or on enhancing the degradation of some special substrates [3]. Main strategies used in remodeling the cell metabolic balance involved introducing heterologous genes, boosting the activity of key genes, relieving the regulation of enzymes, and reducing gene expression of by-product biosynthesis [4–7]. However, genetic modification of C. glutamicum is mainly dependent on the application of antibiotic resistance genes that are used as positive markers for screening target-recombinant strains, especially for the introduction of heterologous genes and overexpression of key genes [8].
Plasmid-mediated hetero-/homologous gene overexpression in amino acid producers has some shortcomings, such as high cost, changes in Corynebacteria’s physiological function, problems in food safety, etc [7, 8]. Although Hu et al. [9] have constructed a novel expression system for gene overexpression using a plasmid system, the antibiotic resistance gene was also introduced into the cell. To bypass the application of the expression plasmid, many researchers have turned to developing integrative plasmids that carry a homologous chromosomal DNA fragment and an antibiotic resistance marker gene [10, 11]. However, the integration involved homologous single-crossover between the plasmid and the recipient genome [8], and the selectable resistance marker was introduced into the cell [10, 11]. In addition, Suzuki et al. [12] have reported a new Cre/mutant lox system for integrating heterologous genes into C. glutamicum genome. However, the host cell must contain a mutant lox gene, and the integrated foreign gene must contain a selective marker, such as antibiotic resistance genes [12]. Therefore, a method that can realize the integration of diverse genes that avoid introducing antibiotic resistance genes is needed.
Previously, we reported on a method to genetically modify the C. glutamicum genome, which involved the integration of diverse genes at a specific gene locus [7]. The method is composed of two recombinant plasmids: the expression plasmid derived from pDXW-8 [13] for cloning the gene cassette, and the mobilizable plasmid derived from pK18mobsacB [14] for targeted gene integration. The recombinant C. glutamicum strains exhibited unmarked gene overexpression and fragment deletion in the genome. However, the recombinant expression plasmid must be constructed every time during targeted gene integration, which was time- and cost consuming. To overcome these problems of heavy workload and high cost, we developed a rapid and efficient method based on a mobilizable plasmid, pK18-MBPMT. The mobilizable plasmid pK18-MBPMT contains RP4 mob, kanamycin (Km) resistance gene kan, genetically modified sacB, promoter P tac, and terminator rrnBT1T2 cartridge with introduced multiple cloning sites (iMCS), derived from pK18mobsacB [14]. This method avoided the construction of expression plasmids, and required only two steps to construct the target integrative plasmids. Therefore, the operation time of this protocol was shorter than the previous method, especially for repeated integration.
As an application example, we demonstrate the locus-specific integration of the heterologous gene amy (encoding α-amylase) and the homologous gene lysC T311I (encoding allosterically feedback-resistant aspartokinase, AKFr) into the C. glutamicum ATCC13032 genome at alaT (encoding aminotransferase T, AlaT) and avtA (encoding alanine-valine aminotransferase, AvtA) loci. The results demonstrated that this method could be used to efficiently modify the genome of C. glutamicum.
Materials and methods
Strains, growth media, and culturing conditions
Strains and plasmids used in this study are listed in Table 1. Luria-Bertani (LB) was used as the standard medium for Escherichia coli. LBG (LB supplemented with 5 g L−1 glucose) was used for C. glutamicum [2]. Epo medium was used for preparing electroporation-competent cells. LB, brain heart infusion, sorbitol (LBHIS) medium was used for obtaining recombinant strains [15]. LBS (LB supplemented with 100 g L−1 of sucrose) and LBK (LB supplemented with 50 µg mL−1 of Km) media were used for screening the recombinant strains [7]. CgXII medium was prepared according to Keilhauer et al. [16]. E. coli and C. glutamicum were grown at 37 and 30 °C, respectively. Where appropriate, strains were cultured with kanamycin (Km 50 µg mL−1), and a reduced concentration of Km (25 µg mL−1) was used to obtain recombinant C. glutamicum.
Construction of mobilizable plasmid pK18-MBPMT
The construction process of plasmid pK18-MBPMT is illustrated in Fig. 1. The expression plasmid pDXW-8 was used as DNA template for amplifying the promoter P tac with iMCS (i.e., ScaI, NotI, XmaIII, XhoI, NdeI, and MluI) and the terminator rrnBT1T2 with the same iMCS by polymerase chain reaction (PCR) using high-fidelity DNA polymerase (Finnzyme, Espoo, Finland) and corresponding primers, respectively (Table 2). It should be pointed out that iMCS was introduced via 5′-extensions of the primers. The PCR fragments were purified by DNA Purification Kit and designated as P tac-iMCS and rrnBT1T2-iMCS, respectively.

Strategy used for the construction of the integrative plasmid pK18-MBPMT. P tac tac promoter, rrnBT1T2 terminator, MCS multiple cloning sites, iMCS introduced multiple cloning sites, P-F/P-R primers for amplifying the promoter P tac fragment, and T-F/T-R primers for amplifying the terminator rrnBT1T2 fragment. The lines indicated in same color represent homologous sequences (color figure online)
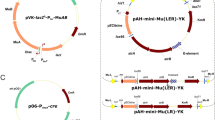
Next, the plasmid pK18mobsacB was digested with BamHI and XbaI and purified by the Gel Extraction Kit. The digested fragments of pK18mobsacB, P tac-iMCS, and rrnBT1T2-iMCS were mixed and supplemented with pEASY ®-Uni Seamless Cloning and Assembly Kit (TRANSGEN, Beijing, China) to assemble these fragments (Fig. 1). After incubation at 50 °C for 15 min, the mixture was electroporated into E. coli JM109 competent cells, and spread onto LBK. The plasmid was extracted from Km-resistant colonies and designated as pK18-MBPMT. The plasmid map is presented in Fig. 2a.

Plasmid maps of a pK18-MBPMT, b pK18-MBPMT/∆alaT::amy, c pK18-MBPMT/∆avtA::lysCT311I, and d pDXW-8-amy
Construction of target integrative plasmid pK18-MBPMT/∆A::B
DNA was extracted from Bacillus amyloliquefaciens or C. glutamicum using the DNA Extraction Kit according to the protocol supplied by the manufacturer (Sangon, Shanghai, China). The construction process of plasmid pK18-MBPMT/∆A::B is shown in Fig. 3. The left and right arms of A gene (representative alaT and avtA) were amplified with high-fidelity DNA polymerase (Finnzyme, Espoo, Finland) from the DNA of C. glutamicum (Table 2) and designated as A-L/A-R (representative alaT-L/alaT-R or avtA-L/avtA-R). The fragments of A-L/A-R were purified, digested with suitable restriction enzymes, respectively (Table 2), and ligated into pK18-MBPMT, which was similarly digested. The resulting plasmid was designated as pK18-MBPMT/∆A (representative pK18-MBPMT/∆alaT or pK18-MBPMT/∆avtA).

Strategy used for the construction of integrative plasmid pK18-MBPMT/∆A::B. A represents knock-out gene (e.g., alaT and avtA); B represents over-expressed gene (e.g., amy and lysC T311I); A-L left arm of A gene, and A-R right arm of A gene, P tac tac promoter; rrnBT1T2 terminator, iMCS introduced multiple cloning sites, A-L-F/A-L-R primers for amplifying the left arm of A gene, and A-R-F/A-R-R primers for amplifying the right arm of A gene, B-F/B-R primers for amplifying the B gene
Next, DNA fragment of B gene (representative amy or lysC T311I genes) was amplified with high-fidelity DNA polymerase (Finnzyme, Espoo, Finland) from the relevant host cell (such as amy from B. amyloliquefaciens CCTCC M2013493 and lysC T311I from C. glutamicum) by corresponding primers (upstream primer contains an SD sequence [17]; Table 2). The fragment was purified, digested with suitable restriction enzymes (Table 2), and ligated into mobilizable plasmid pK18-MBPMT/∆A, which was similarly digested. The resulting plasmid was designated as pK18-MBPMT/∆A::B (representative pK18-MBPMT/∆alaT::amy or pK18-MBPMT/∆avtA::lysC T311I). Plasmids were extracted from E. coli using the Mini Plasmid Kit according to the protocol supplied by the manufacturer (Sangon, Shanghai, China). The plasmid maps are listed in Fig. 2b and c.
Construction of recombinant strains
The plasmid pK18-MBPMT/∆A::B was electroporated into C. glutamicum-competent cells, and the recombinant strains were obtained by screening colonies from Km-resistance and sucrose-sensitivity plates. To clarify the role of this method, we modified amy, lysC T311I, alaT, and avtA in C. glutamicum ATCC 13032. C. glutamicum ATCC 13032 was transformed with plasmid pK18-MBPMT/∆alaT::amy, and the resulted strain was C. glutamicum IA-1. C. glutamicum ATCC 13032 or C. glutamicum IA-1 was transformed with plasmid pK18-MBPMT/∆avtA::lysC T311I, and the resulted strain was C. glutamicum IA-2 or C. glutamicum IAA, respectively. The recombinant strains were screened and confirmed according to Ohnishi et al. [18].
Analytical methods
The cell growth was monitored according to the method as described previously [7]. The glucose and l-lysine concentrations were determined by an SBA-40E immobilized enzyme biosensor (Shandong, China) according to the suggestion of Xu et al. [7]. The cell-free extracts were prepared according to the method as described previously [6]. The cell-free extracts were immediately used for the protein expression analysis by SDS-PAGE [19], and enzyme activities were determined. Protein concentration was determined using the Bradford Protein Quantification Kit according to the protocol supplied by the manufacturer (Sangon, Shanghai, China) using bovine serum albumin as standard. Enzyme activities and protein concentrations were analyzed in biological triplicates. Enzymatic activities of AK [7], AlaT, and AvtA [20] were determined according to the previous reports. The activity of α-amylase was determined by a starch plate test described previously [21] or by Amylase Assay Kit (Biovision, San Francisco, USA) using ethylidene-pNP-G7 as the substrate.
Results
Construction of a mobilizable plasmid pK18-MBPMT
The mobilizable plasmid pK18mobsacB was widely used for gene disruption and exchange by homologous double crossover because of its high performance and absence of genetic markers [14]. Given the composition structure of this plasmid, it cannot be used for gene overexpression. The original pK18mobsacB is composed of MCS, lacZα fragment, kan, genetically modified sacB, etc., but is devoid of promoter and terminator [14]. The constructed plasmid pK18-MBPMT contained not only the elements of pK18mobsacB, but also a P tac-iMCS-rrnBT1T2 cartridge (Fig. 2a). The general scheme for constructing pK18-MBPMT based on pK18mobsacB was shown in Fig. 1. A 319-bp long fragment contained with homologous sequences upstream of the BamHI site in pK18mobsacB, promoter P tac and iMCS, a 270-bp long fragment contained with iMCS, terminator rrnBT1T2 and homologous sequences downstream of the XbaI site in pK18mobsacB, and the plasmid pK18mobsacB digested with BamHI and XbaI were ligated using the pEASY ®-Uni Seamless Cloning and Assembly Kit. Successful constructions were selected by plating transformed E. coli JM109 on LBK. The target plasmid was extracted from E. coli and examined by the restriction endonuclease analysis (data not shown). The target mobilizable plasmid was selected and designated as pK18-MBPMT.
Construction of a target integrative plasmid pK18-MBPMT/∆A::B
In contrast to our previous report [7], there were two steps to construct pK18-MBPMT/∆A::B (representative pK18-MBPMT/∆alaT::amy or pK18-MBPMT/∆avtA::lysCT311I) (Fig. 3): (1) construction of pK18-MBPMT/∆A carrying the left and right arms of A gene, which provided homology for recombination and (2) insertion of B gene into pK18-MBPMT/∆A, the resulting plasmid designated as pK18-MBPMT/∆A::B. Since the plasmid pK18-MBPMT contained the P tac and rrnBT1T2 (Fig. 2a), the construction of pDXW-8-B was not required to obtain the cassette P tac-B-rrnBT1T2. Moreover, there was an iMCS between P tac and rrnBT1T2 (Fig. 2a), which was beneficial to allow direct insertion of B gene into plasmid pK18-MBPMT/∆A. All procedures were performed within 48 h, whereas our previous method required at least 72 h [7].
Integration of heterologous α-amylase gene in C. glutamicum genome
A heterologous α-amylase-encoding gene, amy from B. amyloliquefaciens was used to confirm the potential of mobilizable plasmid pK18-MBPMT-mediated DNA integration approach in C. glutamicum. To this end, the plasmid pK18-MBPMT/∆alaT::amy (Fig. 2b) was introduced into C. glutamicum ATCC 13032 by electroporation (see “Materials and methods”). The target-recombinant strains were obtained according to Xu et al. [7]. The colonies were randomly selected from LBS plates and confirmed by individual bacterial colonies PCR, indicating that 92 % of the resulting colonies contained the amy gene (data not shown). We randomly selected one colony and sequenced it via Sangon Biotech (Shanghai) Co., Ltd. (Shanghai, China) to further reduce the false positive, and the resulted strain was designated as C. glutamicum IA-1. To confirm whether the integration happened at alaT locus, PCR was performed using the alaT-L-F/alaT-R-R primer pair, and approximately 3.8- and 2.5-kbp DNA fragments were amplified from C. glutamicum IA-1 and the wild-type strain C. glutamicum ATCC13032, respectively (Fig. 4b). Moreover, to confirm the normal expression of integrated amy in C. glutamicum genome, α-amylase activity was determined and compared to the expression plasmid pDXW-8-amy (Fig. 2d) mediating amy gene expression. As shown in Fig. 4a, C. glutamicum IA-1 possessed the capability of hydrolyzing starch at about the same level as the strain harboring pDXW-8-amy. These results were consistent with results obtained from the Amylase Assay Kit (Table 3). The α-amylase activity of C. glutamicum IA-1 was 670 ± 2.4 mU (mg protein)−1, while C. glutamicum/amy showed 840 ± 9.1 mU mg−1. In addition, cell-free extracts prepared from the above-mentioned strains were analyzed using SDS-PAGE (Fig. 4c). The thick protein band observed in the cell-free extracts from C. glutamicum IA-1 and C. glutamicum/amy but not in C. glutamicum ATCC13032 was consistent with the expected molecular mass of α-amylase (~56 kDa) (Fig. 4c). These results indicated that the expression of α-amylase by the C. glutamicum strains was either via integration into genome or plasmid-mediated expression.

Site-directed integration of amy in C. glutamicum ATCC13032. a Iodine staining of 1 % starch-containing agar plates after inoculation with wild-type strain and recombinant strains. Clear zones indicate hydrolysis of starch around recombinant colonies. b PCR amplification of wild-type strain and strain IA-1 with alaT-L-F/alaT-R-R (Lanes 1 and 3) or amy-F/amy-R (Lanes 2 and 4) as primer pairs. M is the DNA marker (DL15000). c SDS-PAGE analysis of α-amylase expression in C. glutamicum ATCC13032. Lane 1 C. glutamicum ATCC13032; Lane 2 C. glutamicum/amy; Lane 3 C. glutamicum ATCC13032 harboring pDXW-8; Lane 4 C. glutamicum IA-1; Lane 5 C. glutamicum IAA; M is the protein marker. Arrow indicates bands corresponding to α-amylase protein
Integration of homologous AKFr gene in C. glutamicum genome
To further demonstrate the feasibility of this strategy for integrating homologous gene into C. glutamicum genome, AKFr-encoding gene lysC T311I from C. glutamicum was integrated into C. glutamicum genome. To achieve this, the pK18-MBPMT/∆avtA::lysCT311I (Fig. 2c) was introduced into C. glutamicum ATCC 13032 or C. glutamicum IA-1 by electroporation. Colonies were randomly selected from LBS plates and confirmed by colony PCR using avtA-L-F/avtA-R-R as primer pair, indicating that about 90 % of the resulting colonies contained the desired mutations (Fig. 5a). The target-recombinant strains were designated as C. glutamicum IA-2 or C. glutamicum IAA, respectively. The expected chromosomal localization of integrated pK18-MBPMT/∆avtA::lysCT311I (Fig. 5a) was confirmed by amplifying a specific 3.7-kbp DNA fragment from recombinant strains by PCR using primer pair avtA-L-F/avtA-R-R, whereas the fragment from strain ATCC13032 and IA-1 was about 2.4 kbp (Fig. 5a). In addition, AK activity of C. glutamicum IA-2 and C. glutamicum IAA was higher than that of the wild-type strain ATCC13032 and strain IA-1 (Table 3). Moreover, AvtA activity of strain IA-2 and IAA was not detected (Table 3). As described previously, the inactivation of alaT and avtA led to the recombinant strain unable to grow in CgXII medium containing glucose unless supplemented with l-alanine (Fig. 5b). These results indicated that lysC T311I was inserted into C. glutamicum genome at the avtA locus and expressed successfully.

Site-directed integration of lysC T311I in C. glutamicum ATCC13032 and C. glutamicum IA-1. a PCR amplification of wild-type strain and recombinant strains with avtA-L-F/avtA-R-R as primer pairs. Lane 1–10 indicates site-directed integration of lysC T311I in C. glutamicum ATCC13032; Lane 11–21 indicates site-directed integration of lysC T311I in C. glutamicum IA-1. M is the DNA marker (DL15000). b Growth of wild-type strain and recombinant strains on CgXII medium containing 40 g L−1 glucose (−) or supplemented with 1 g L−1 l-alanine (+); 13032 indicates C. glutamicum ATCC13032
l-Lysine production of genetically defined C. glutamicum strains
The results above indicated that the integrated lysC T311I could be successfully expressed in C. glutamicum, and the activities of AlaT and AvtA were successfully inactivated (Table 3). However, lysC T311I, alaT, and avtA have important roles in regulating l-lysine production by C. glutamicum. To investigate whether integration or inactivation of these genes via this method was beneficial to the improvement of l-lysine production, all strains were grown in CgXII minimal medium containing 40 g L−1 glucose and/or 1 g L−1 l-alanine, and growth and l-lysine accumulation were monitored over the course of the experiment. The recombinant strains IA-1, IA-2, and IAA showed slightly lower cell growth than wild-type strain ATCC13032 (Table 4). However, l-lysine production in the recombinant strains IA-2 and IAA was significantly increased: 78.0 ± 1.76 mmol mol−1 (i.e., C-mmol l-lysine/C-mol glucose) and 102.6 ± 3.15 mmol mol−1, respectively, whereas l-lysine was not detected in strains ATCC13032 and IA-1 (Table 4). In addition, the biomass-specific l-lysine yield (Y P/X) of strains IA-2 and IAA was also significantly increased (Table 4).
Discussion
Plasmid-mediated gene overexpression in Corynebacterium is a common strategy in genetic engineering [7]. However, gene overexpression via plasmid is not beneficial to cell growth because of metabolic imbalance and accumulation of intermediates [8]. In addition, antibiotics must be added to the broth to maintain plasmid stability, which will increase production cost for companies and causes worry among the public [7, 22]. Therefore, many researchers dedicate into the research of DNA integration techniques, because it is the best genetic tool to stably express desirable genes in bacteria [12]. C. glutamicum shows complicated restriction systems (e.g., a specific DNA methylation pattern) that can severely affect the integration of foreign gene into its genome [12, 23]. To date, despite the development of many methods used to integrate foreign genes into the C. glutamicum genome, most of them will also introduce antibiotic resistance genes into the host cell [9–12]. In addition, homologous single-crossover between integrated plasmid and genome is the main strategy in these methods, which has low integration efficiency [8]. To increase the efficiency of integration, Vertès et al. [24] developed a new method, which depended on homologous double crossover. Although the Km-resistance gene was introduced into the host cell, the integration frequency of target insertion reached 2.4 × 102 integrants (μg DNA)−1 [24]. Therefore, methods based on homologous double crossover might be useful to increase the integration efficiency.
The mobilizable plasmid pK18mobsacB is widely used for gene disruption and allelic exchange by homologous double crossover without genetic markers and showed high performance [14]. This plasmid is mainly used in gene disruption and exchange, but it is seldom used to gene overexpression [14, 18, 25]. Previously, we confirmed that it could work well in modifying the C. glutamicum genome to implement gene overexpression [7]. To overcome the heavy workload and high-cost of the previous method [7], a method based on mobilizable plasmid pK18-MBPMT has been developed. The mobilizable plasmid pK18-MBPMT contains promoter P tac, terminator rrnBT1T2, and iMCS except for the components of pK18mobsacB. This method avoids the construction of expression plasmids, and only needs two steps to construct the target integrative plasmids. Using this method, the operation time of this protocol was shorter than the previous method (2 vs. 3 days), especially for repeated integration.
We showed the feasibility of the present strategy by integrating pK18-MBPMT/∆alaT::amy harboring the amy gene from B. amyloliquefaciens, which provided C. glutamicum ATCC13032 with functional α-amylase activity. Normally, C. glutamicum does not possess functional α-amylase activity in media [24], but recombinant strains integrated with the B. amyloliquefaciens amy can break down soluble starch, indicating that the heterologous amy gene can be successfully expressed in C. glutamicum (Fig. 4a). To further demonstrate the potential of this strategy for integrating a homologous gene into the C. glutamicum genome, the lysC T311I gene from C. glutamicum was integrated into the genome at the avtA locus. The lysC T311I gene, encoding AKFr, is a key enzyme in l-lysine biosynthetic pathway [18]. The alaT and avtA, encoding aminotransferase T and A, respectively, are necessary for l-alanine biosynthesis [2]. Overexpression of lysC T311I proved to beneficially improve l-lysine production [4], whereas the inactivation of alaT or avtA showed a decrease in l-alanine accumulation [2, 4]. Therefore, the increased l-lysine production indicated that lysC T311I has been successfully integrated into the C. glutamicum genome and expressed via plasmid pK18-MBPMT/∆avtA::lysCT311I. In addition, integrating amy and lysC T311I at alaT and avtA loci, respectively, in strain C. glutamicum IAA resulted in its inability to grow in CgXII medium containing glucose unless supplemented with l-alanine (Fig. 5b). This indicated that avtA and alaT were successfully inactivated. These results are in accordance with the previous results [2, 4], indicating that the gene unmarked site-specific integration and fragment deletion in C. glutamicum genome using mobilizable plasmid pK18-MBPMT was successfully applied in improving l-lysine production.
In conclusion, genetically modifying the genome is an important strategy to stably express desirable genes in bacteria. The mobilizable plasmid pK18-MBPMT-mediated gene integration is useful for the improvement of the C. glutamicum genome. It could even create a C. glutamicum minimum genome factory (MGF), because this method can achieve gene overexpression and inactivation at the same time, and the genetically modified strains do not contain any genetic markers. In addition, the operation time of this protocol is shorter than the previous method, especially for repeated integration. Therefore, we believe the method described in this study has promising applications in genetically modifying industrial strains for high-level amino acid production.
Abbreviations
- AKFr :
-
Allosterically feedback-resistant aspartokinase
- AlaT:
-
Aspartate aminotransferase
- AvtA:
-
PLP-dependent aminotransferase
- iMCS:
-
Introduced multiple cloning sites
- Km:
-
Kanamycin
- DCW:
-
Dry cell weight
- μ max :
-
Maximum specific growth rate
- LB:
-
Luria-Bertani
- LBG:
-
LB + 5 g L−1 glucose
- LBS:
-
LB + 100 g L−1 sucrose
- LBK:
-
LB + 50 µg mL−1 Km
- LBHIS:
-
LB + brain heart infusion + sorbitol
References
Wieschalka S, Blombach B, Bott M, Eikmanns BJ (2013) Bio-based production of organic acids with Corynebacterium glutamicum. Microb Biotechnol 6:87–102. doi:10.1111/1751-7915.12013
Hou XH, Chen XD, Zhang Y, Qian H, Zhang WG (2012) L-Valine production with minimization of by-products’ synthesis in Corynebacterium glutamicum and Brevibacterium flavum. Amino Acids 43:2301–2311. doi:10.1007/s00726-012-1308-9
Pátek M, Nešvera J, Guyonvarch A, Reyes O, Leblon C (2003) Promoters of Corynebacterium glutamicum. J Biotechnol 104:311–323. doi:10.1016/j.jbiotec.2005.12.027
Becker J, Zelder O, Häfner S, Schröder H, Wittmann C (2011) From zero to hero-design-based systems metabolic engineering of Corynebacterium glutamicum for l-lysine production. Metab Eng 13:159–168. doi:10.1016/j.ymben.2011.01.003
van Ooyen J, Noack S, Bott M, Reth A, Eggeling L (2012) Improved l-lysine production with Corynebacterium glutamicum and systemic insight into citrate synthase flux and activity. Biotechnol Bioeng 109:2070–2081. doi:10.1002/bit.24486
Xu JZ, Han M, Zhang JL, Guo YF, Zhang WG (2014) Metabolic engineering Corynebacterium glutamicum for the l-lysine production by increasing the flux into l-lysine biosynthetic pathway. Amino Acids 46:2165–2175. doi:10.1007/s00726-014-1768-1
Xu JZ, Xia XH, Zhang JL, Guo YF, Zhang WG (2014) A method for gene amplification and simultaneous deletion in Corynebacterium glutamicum genome without any genetic markers. Plasmid 72:9–17. doi:10.1016/j.plasmid.2014.02.001
Ikeda M, Katsumata R (1998) A novel system with positive selection for the chromosomal integration of replicative plasmid DNA in Corynebacterium glutamicum. Microbiol 144:1863–1868
Hu JY, Li YY, Zhang HL, Tan YZ, Wang XY (2014) Construction of a novel expression system for use in Corynebacterium glutamicum. Plasmid 75:18–26. doi:10.1016/j.plasmid.2014.07.005
Correia A, Martin JF, Castro JM (1996) Targeted integration of foreign genes into repetitive sequences of the Brevibacterium lactofermentum genome. FEMS Microbiol Lett 142:259–264. doi:10.1111/j.1574-6968.1996.tb08440.x
Amador E, Franciso J, José M, Castro M (2007) A Brevibacterium lactofermentum 16S rRNA gene used as target site for homologous recombination. FEMS Microbiol Lett 185:199–204. doi:10.1111/j.1574-6968.2000.tb09062.x
Suzuki N, Inui M, Yukawa H (2007) Site-directed integration system using a combination of mutant lox sites for Corynebacterium glutamicum. Appl Microbiol Biotechno 77:871–878. doi:10.1007/s00253-007-1215-2
Xu DQ, Tan YZ, Huan XJ, Hu XQ, Wang XY (2010) Construction of a novel shuttle vector for use in Brevibacterium flavum, an industrial amino acid producer. J Microbiol Methods 80:86–92. doi:10.1016/j.mimet.2009.11.003
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutumicum. Gene 145:69–73. doi:10.1016/0378-1119(94)90324-7
van der Rest ME, Lange C, Molenaar D (1999) A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol 52:541–545. doi:10.1007/s002530051557
Keilhauer C, Eggeling L, Sahm H (1993) Isoleucine synthesis in Corynebacterium glutamicum: molecular analysis of the ilvB-ilvN-ilvC operon. J Bacteriol 175:5595–5603
Srivastava P, Deb JK (2005) Gene expression systems in corynebacteria. Protein Expression Purif 40:221–229. doi:10.1016/j.pep.2004.06.017
Ohnishi J, Mitsuhashi S, Hayashi M, Ando S, Yokoi H, Ochiai K, Ikeda M (2002) A novel methodology employing Corynebacterium glutamicum genome information to generate a new l-lysine-producing mutant. Appl Microbiol Biotechnol 58:217–223. doi:10.1007/s00253-001-0883-6
Xu JZ, Zhang JL, Guo YF, Zhang WG (2015) Genetically modifying aspartate aminotransferase and aspartate ammonia-lyase affects metabolite accumulation in l-lysine producing strain derived from Corynebacterium glutamicum ATCC13032. J Mol Catal B Enzym 113:82–89. doi:10.1016/j.molcatb.2014.12.015
Marienhagen J, Eggeling L (2008) Metabolic function of Corynebacterium glutamicum aminotransferases AlaT and AvtA and impact on l-valine production. Appl Environ Microbiol 74:7457–7462. doi:10.1128/AEM.01025-08
Ugorčáková J, Jucovič M, Bukovská G, Timko J (1996) Construction and characterization of new corynebacterial plasmids carrying the α-amylase gene. Folia Microbiol 41:10–14. doi:10.1007/BF02816332
Tauch A, Götker S, Pühler A, Kalinowski J, Thierbach G (2002) The alanine racemase gene alr is an alternative to antibiotic resistance genes in cloning systems for industrial Corynebacterium glutamicum strains. J Biotechnol 99:79–91. doi:10.1016/S0168-1656(02)00159-1
Schäfer A, Tauch A, Droste N, Pühler A, Kalinowski J (1997) The Corynebacterium glutamicum cglIM gene encoding a 5-cytosine methyltransferase enzyme confers a specific DNA methylation pattern in an McrBC-deficient Escherichia coli strain. Gene 203:95–101. doi:10.1016/S0378-1119(97)00519-2
Vertès AA, Hatakeyama K, Inui M, Kobayshi M, Kurusu Y, Yukawa H (1993) Replacement recombination in Coryneform bacteria: high efficiency integration requirement for non-methylated plasmid DNA. Biosci Biotechnol Biochem 57:2036–2038. doi:10.1271/bbb.57.2036
Vogt M, Haas S, Klaffl S, Polen T, Eggeling L, van Ooyen J, Bott M (2014) Pushing product formation to its limit: metabolic engineering of Corynebacterium glutamicum for l-leucine overproduction. Metab Eng 22:40–52. doi:10.1016/j.ymben.2013.12.001
Kinoshita S, Udaka S, Shimono M (1957) Studies on the amino acid fermentation Part I. production of l-glutamic acid by various microorganisms. J Gen Microbiol 3:193–205
Acknowledgments
This work was financially supported by the Youth Program of Natural Science Foundation of Jiangsu Province (No. BK20150149) and the Youth Foundation of Jiangnan University (No. JUSRP115A19).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xu, J., Zhang, J., Han, M. et al. A method for simultaneous gene overexpression and inactivation in the Corynebacterium glutamicum genome. J Ind Microbiol Biotechnol 43, 1417–1427 (2016). https://doi.org/10.1007/s10295-016-1806-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-016-1806-y