
Abstract
The genus Streptomyces comprises bacteria that undergo a complex developmental life cycle and produce many metabolites of importance to industry and medicine. Streptomyces clavuligerus produces the β-lactamase inhibitor clavulanic acid, which is used in combination with β-lactam antibiotics to treat certain β-lactam resistant bacterial infections. Many aspects of how clavulanic acid production is globally regulated in S. clavuligerus still remains unknown. We conducted comparative proteomics analysis using the wild type strain of S. clavuligerus and two mutants (ΔbldA and ΔbldG), which are defective in global regulators and vary in their ability to produce clavulanic acid. Approximately 33.5 % of the predicted S. clavuligerus proteome was detected and 192 known or putative regulatory proteins showed statistically differential expression levels in pairwise comparisons. Interestingly, the expression of many proteins whose corresponding genes contain TTA codons (predicted to require the bldA tRNA for translation) was unaffected in the bldA mutant.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Streptomyces are Gram-positive, soil-dwelling bacteria that have a complex life cycle involving vegetative mycelia, aerial hyphae and unigenomic spores [44, 57]. The vegetative mycelia grow into the substrate to obtain nutrients and give rise to aerial hyphae, which are responsible for the characteristic “fuzzy” appearance of Streptomyces colonies. Over time, the aerial hyphae undergo septation to produce pigmented spores that assist in dispersal [1, 2]. The Streptomyces are also known for their ability to produce a large variety of secondary metabolites, many of which have applications in medicine, agriculture and industry. Streptomyces clavuligerus is capable of producing a number of secondary metabolites which include the β-lactams: clavulanic acid (CA), cephamycin C, and a group of metabolites collectively referred to as the 5S clavams [16]. CA has potent β-lactamase inhibitory activity and is used in the treatment of bacterial infections that are otherwise resistant to certain β-lactam antibiotics [77]. CA and the 5S clavams partially share a common ‘early’ biosynthetic pathway and have dedicated ‘late’ steps involved in the production of the respective metabolites [41, 91]. In addition, multiple gene clusters have been shown to be involved in the production of CA and the 5S clavams, with the primary CA biosynthetic gene cluster being located adjacent to the cephamycin C gene cluster in S. clavuligerus [104]. Currently, CA is produced at the industrial level by fermenting S. clavuligerus as total chemical synthesis on a large scale is not economically or technically feasible [7].
The expression of many genes involved in secondary metabolism in the Streptomyces is regulated in a hierarchical manner [8, 66]. At the first level of control are the pathway-specific (or cluster-situated) transcriptional regulators that are encoded by genes located within or in the vicinity of the gene clusters that they regulate [101]. These regulators are in turn controlled by pleiotropic or global regulatory elements that coordinate secondary metabolism and morphological development [8, 66]. Two pathway-specific regulators are of particular importance during CA biosynthesis. The claR gene from the CA gene cluster encodes a LysR-family transcriptional regulator, which controls the expression of the ‘late’ genes involved exclusively in CA biosynthesis [67]. Recently, it was also reported that the expression of other genes involved in development and secondary metabolism is also affected in a S. clavuligerus claR mutant [56]. The ccaR gene from the cephamycin C gene cluster encodes a member of the Streptomyces antibiotic regulatory protein (SARP) family and is required for the production of cephamycin C and CA, but not for that of the 5S clavams [2]. CcaR regulates CA production both directly and indirectly by controlling the expression of the “early” genes from the CA gene cluster [94] and by regulating the expression of claR [67, 70, 82, 83], respectively.
Pleiotropic regulators controlling secondary metabolism and/or morphological development in S. clavuligerus include the products of the bldA and bldG genes, which encode a tRNA and an anti–anti-sigma factor (AASF), respectively [12, 48]. bldA and bldG are highly conserved among Streptomyces spp. and are part of the bld (bald) gene family, which get their name from the fact that mutations in these genes lead to loss of aerial hyphae formation and result in smooth colonies instead of the characteristic fuzzy appearance of wild type (wt) colonies [11, 24, 48, 61, 63, 73]. In S. coelicolor and S. griseus, the BldG AASF is known to interact with different anti-sigma factor (ASF) proteins to regulate multiple sigma factors by a partner-switching mechanism [60, 69, 86, 96]. Deletion of bldG completely abolishes the production of secondary metabolites and aerial hyphae in S. clavuligerus [11]. The loss of CA and cephamycin C production in the S. clavuligerus bldG mutant is due to blocked expression of ccaR, although at this time it is still unclear as to how BldG regulates ccaR transcription [11]. The bldA gene encodes a leucine-specific tRNA required for translating TTA codons, which is the rarest codon present in Streptomyces genes due to the high GC content of their DNA [33]. Mutations in bldA result in the loss of aerial hyphae formation and secondary metabolite production in S. coelicolor, S. lividans and S. griseus [47, 48], whereas an S. clavuligerus bldA mutant is developmentally defective but can still produce CA and cephamycin C at reduced levels as compared to the wt strain [97]. This finding was initially surprising as ccaR contains a TTA codon, but the production of the CcaR protein still occurred in a S. clavuligerus bldA mutant, suggesting that the TTA codon in ccaR is somehow mistranslated using another tRNA [97].
BldG is known to regulate CA and cephamycin C production and morphological differentiation in S. clavuligerus [11] and has been shown to interact with multiple proteins in other Streptomyces spp. [60, 69, 86]. In contrast, the bldA tRNA is required for proper morphological development but not CA and cephamycin C production in S. clavuligerus [97]. Therefore, quantitative proteome-wide changes in the S. clavuligerus bldG and bldA mutants were examined using isobaric tag for relative and absolute quantitation (iTRAQ) for comparison to the wt strain. This was done to obtain a global survey of protein expression profiles in the three strains and to attempt to differentiate between proteins involved in secondary metabolism and morphological differentiation. The study was further focused to identify proteins known or predicted to be involved in regulation to identify potential elements involved in controlling CA production in S. clavuligerus at a level above that of CcaR.
Materials and methods
Bacterial strains, culture conditions and detection of CA in supernatants
Streptomyces clavuligerus NRRL3585 strains were maintained on International Streptomyces Project medium 4 (ISP-4) or maltose-yeast extract-malt extract (MYM) agar media [46]. Ten ml of trypticase soy broth seed cultures were grown for 48 h in 50 ml flasks supplemented with 2 % w/v starch (TSBS). Two ml of the seed cultures were used to inoculate 100 ml of SA production cultures in 500 ml flasks. All S. clavuligerus cultures were grown at 28 °C and liquid cultures were agitated using a rotary shaker at 150 rpm. Cultures used for subsequent proteomics analysis were also monitored by Gram staining and microscopy to verify growth characteristics and purity. Standard antibiotic concentrations were used when required [11]. Bioassays to detect CA production in culture supernatants were performed using previously described procedures [94]. Reversed phase high performance liquid chromatography (HPLC) analysis of imidazole-derivatized culture supernatants was performed using a 100 × 8 mm Bondclone™ 10 µm C18 148 Å LC Column (Phenomenex Inc., USA) as described previously [31]. All media and reagents were purchased from Fisher Scientific or VWR International (Canada) unless otherwise specified.
Sample preparation, iTRAQ labeling and LC–MS/MS analysis
One hundred ml of SA cultures grown for 48 h were harvested by centrifugation at room temperature and the pellets were re-suspended in 3 ml phosphate buffered saline (pH 7.4) containing 0.01 % w/v SDS. The cells were disrupted immediately by sonication on ice, followed by centrifugation and the supernatants were frozen in liquid N2 for subsequent analysis. Preliminary quantification of protein concentrations for growth determination was carried out using the OmniPur™ BRADFORD Kit (EMD/Millipore, USA). All subsequent proteomics analysis was carried out at the Genome BC Proteomics Centre, University of Victoria, Victoria, BC, Canada. Protein samples were quantified and precipitated overnight in acetone followed by resolubilization in triethylammonium bicarbonate (TEAB) and 0.2 % w/v SDS. Samples were reduced with tris(2-carboxyethyl)phosphine hydrochloride (TCEP), alkylated with methyl methanethiosulfonate (MMTS), digested in-solution with trypsin and labeled with the appropriate 8-plex iTRAQ label according to the manufacturers recommendations (Applied Biosystems). Different labels used for each extract were as follows: wt (113, 114, and 115), ΔbldG (116, 117, and 118) and ΔbldA (119 and 121).
The iTRAQ labeled peptides were combined, desalted, and concentrated on an Oasis HLB cartridge (Waters Corp.), and speed vacuum centrifuged to dryness before separation by high pH reversed phase HPLC. Fractions were collected every 30 s for 96 min and were concatenated into 24 fractions by combining every 24th fraction (e.g., fractions 1, 25, 49, 73 were combined). The length of the reverse gradient was 2 h per HPLC fraction. Each of the samples was rehydrated to 110 μl (2 % acetonitrile, 0.1 % formic acid). Following rehydration, 15 μl aliquots of the peptide solutions were separated by on-line reversed phase liquid chromatography using a Thermo Scientific EASY-nLCII system with a reversed phase Magic C-18AQ (75 μm ID, 2 cm length, 5 μm, 100 Å, Michrom BioResources, Inc.) pre-column and a Magic C-18AQ nano-analytical column (75 µm ID, 15 cm length, 5 µm, 100 Å, Michrom BioResources, Inc.) at a flow rate of 300 nl/min. The chromatography system was coupled on-line to an LTQ Orbitrap Velos mass spectrometer equipped with a Nanospray Flex source (Thermo Scientific). The solvents used are as follows: A 2 % acetonitrile, 0.1 % formic acid; B 90 % acetonitrile, 0.1 % formic acid. After a 249 bar (~5 μl) pre-column equilibration and a 249 bar (~8 μl) nano-column equilibration, samples were separated using a 120 min gradient (0 min 5 % B; 100 min 40 % B; 5 min 80 % B; 2 min 95 % B; 13 min 95 % B). The parameters for the LTQ Orbitrap Velos (Thermo Scientific) were as follows: Nano-electrospray ion source with spray voltage 2.1 kV; capillary temperature 250 °C; Survey MS1 scan m/z range 400–1800 profile mode; resolution 30,000 at m/z 400 with AGC target 1E6; and one microscan with maximum inject time 500 ms. A siloxane m/z 445.120024 lock mass was used for internal calibration. Other settings were: preview mode for FTMS master scans: on; injection waveforms: on; monoisotopic precursor selection: on; rejection of charge state: 1. The experiment was acquired as a top ten data dependent analysis of the most abundant ions with charge states of 2–4, exceeding 5000 counts, being selected for higher-energy collisional dissociation (HCD) FT MS/MS fragmentation (scans 2–11) and detection in centroid mode. Dynamic exclusion settings were: repeat count 1; repeat duration 15 s; exclusion list size 500; exclusion duration 15 s with a 10 ppm mass window. The FT HCD settings were: resolution 7500 at m/z 400 in centroid mode with AGC target 1E5, amu isolation width, and normalized collision energy 50 %, 0.1 ms activation time.
Data analysis
The raw files generated from LC–MS/MS were analyzed using Proteome Discoverer 4.1 software suite (Thermo Scientific). Spectrum Selection was used to generate peak lists of the HCD spectra (parameters: activation type HCD; s/n cut-off 1.5; total intensity threshold 0; minimum peak count 1; precursor mass 350–5000 Da). A list of S. clavuligerus proteins was created using the two available S. clavuligerus full genome sequences (NZ_ADWJ01000000 and NZ_ADGD01000000) as well as the four plasmids (NZ_CM001016.1, NZ_CM001017.1, NZ_CM001018.1, and NZ_CM001019.1) using Artemis (Wellcome Trust Sanger Institute). The sequences of all predicted S. clavuligerus proteins were uploaded to the Mascot 2.4 server and the and the peak list generated by Proteome Discoverer 4.1 was used to search the database using the following parameters: precursor tolerance 10 ppm; MS/MS tolerance 20 mmu for FT MS/MS HCD data; enzyme Trypsin; two missed cleavages; instrument type FT-ICR ESI; fixed modification: Methylthio (C), iTRAQ 8-plex (K), and iTRAQ 8-plex (N-term); variable modifications: Oxidation (M), Deamidated (NQ), iTRAQ® 8-plex (Y) [18]. The HCD Percolator settings were: max delta Cn 0.05; target false discovery rate (FDR) strict 0.01, target FDR relaxed 0.05 with validation based on q value [18].
36,780 peptides with a single S. clavuligerus protein database match from the Mascot search were retained for further analysis. To ensure that only data that were included by Proteome Discoverer during quantitation analysis were used, peptides had to be classified as “used,” which requires the presence of an iTRAQ label and at least one valid iTRAQ reporter ion ratio. The median abundance of multiple peptides uniquely mapped to a single protein was used as the abundance measurement for that protein. All protein abundance measurements that were missing or were <1, were set to 1 and the measurements were log2 transformed. Replicates for each strain were normalized together using quantile normalization available in the normalize between arrays function in the R package, Limma (version 3.16.5). Pairwise differential protein expression between the three strains (i.e., ΔbldG–wt, ΔbldA–wt, ΔbldA–ΔbldG) was identified by means of a moderated paired t test [88] available in the Bioconductor package Limma. P values were corrected for multiple testing using the false discovery rate (FDR) approach [5]. Proteins with a FDR-corrected P value of <0.01 in any given comparison were deemed to have a statistically significant differential expression in that comparison.
Functional annotations for S. clavuligerus proteins were obtained by searching the protein sequences against Pfam database [29], NCBI Conserved Domain Database (CDD) [54] and dcGO database [26]. Based on manual examination of these functional annotations and on the genomic location of the respective encoding genes, proteins were classified in one of five functional groups; namely, metabolism, cellular processes, regulation, energetics and translation. Number of proteins per functional group differentially expressed in the ΔbldG–wt, ΔbldA–ΔbldG, ΔbldA–wt comparisons were counted and displayed using the barplot2 function in the R package gplots (version 2.14.1). Proteins found differentially expressed in more than one comparison were counted only once taking into account the comparisons in the following order: ΔbldG/wt, ΔbldA/ΔbldG and ΔbldA/wt. In addition, differentially expressed regulatory proteins (±2 fold and more) were identified for further analysis.
Z scores were calculated with the formula (x − μ)/σ where x is the normalized abundance of a protein in a sample, μ is the average abundance of that protein across all samples and σ is the standard deviation of the same protein across all samples. Heat maps of Z scores of proteins involved in regulation were obtained using the function heatmap.2 available in the R package gplots. The function was executed using hierarchical clustering with complete linkage and Euclidean distance.
Identification of TTA codons
The full genome sequence data of the S. clavuligerus chromosome (NZ_CM001015), as well as the four plasmids, pSCL1 (NZ_CM001016), pSCL2 (NZ_CM001017), pSCL3 (NZ_CM001018), and pSCL4 (NZ_CM001019), were obtained in FASTA format from NCBI as described above, and was uploaded to the TTA Lynx server which filtered out all genes containing TTA codons [109]. The proteins encoded by these genes were then manually searched for in the data set. The location and context of each TTA codon was also verified manually.
Results
Sampling conditions and summary of proteomics analysis
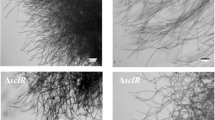
A survey of CA production was first carried out to verify that the strains being subjected to detailed proteomics analysis displayed previously reported phenotypes [11, 97] in terms of metabolite production when grown in starch asparagine (SA) medium. SA was selected as it is a defined medium that supports the production of CA but not of the 5S clavams [42], making it a good choice for obtaining samples for proteomic profiles specifically during CA biosynthesis in S. clavuligerus. Bioassays and HPLC analysis of culture supernatants from the three strains grown for up to 120 h were performed using samples taken at 24 h intervals (Fig. 1). As expected, CA was detected in culture supernatants from the wt strain and the bldA mutant at all of the time points examined, whereas no CA was detected in the bldG mutant samples at any of the time points (Fig. 1a). Although CA production still occurred in the bldA mutant, quantitative analysis indicated that production was reduced by 80 % in this strain as compared to wt (Fig. 1b).

Clavulanic acid production and growth of the S. clavuligerus wt, ΔbldA and ΔbldG strains grown in SA medium. a Bioassay results for the respective strains grown for up 120 h. b Quantitative HPLC results for clavulanic acid production levels using imidazole-derivatized supernatants from the respective strains after 48 h of growth. c Growth levels of the three strains at the 48 h time point, which was used for harvesting mycelia for subsequent proteomics analysis. d Morphologies of mycelia from the three strains at the 48-h time point showing growth in the form of mycelial mats
Previous studies have reported that CA production in wt S. clavuligerus peaks after approximately 96 h of growth in SA and in other media [68]. Unlike many other secondary metabolite producers, the production of cephamycin C and CA in S. clavuligerus takes place during exponential growth and is not initiated during idiophase [81]. Therefore, the 48 h time point during growth in SA was chosen for sampling to obtain a proteomic profile at the onset of CA production based on our results and previous reports [3, 93]. The growth of all strains was comparable in SA media at the 48-h time point (Fig. 1c) and took place in the form of mycelial mats (Fig. 1d). A list of the amino acid sequences of all proteins predicted to be encoded by the S. clavuligerus chromosome and plasmids was compiled based on previously reported genome sequences (Supplementary File 1) [58, 92], and was used in the analysis. A total of 2496 S. clavuligerus proteins were detected in the subsequent iTRAQ analysis of which 2442 met the strict confidence criteria as described in the methods section, and were therefore included in the study (Supplementary File 2). This corresponds to approximately 33.5 % of the entire S. clavuligerus proteome (Supplementary File 1), as the S. clavuligerus genome has been predicated to encode 7281 proteins [58, 92].
Data normalization and differential expression analysis
Quantile normalization of the replicates from each strain was performed to correct for variations in protein abundance [13]. Analysis showed that replicates from each strain clustered together, demonstrating that protein expression levels between the replicates was comparable (Supplementary File 3). Therefore, the quantile normalized data was used for further analysis. Pair-wise differential protein expression patterns between strains were determined using a moderated paired t test [78] with FDR-corrected P values, in which P values of <0.01 in any given comparison were deemed to be statistically significant (Supplementary File 4). Further analysis showed that 275 proteins were over-expressed and 641 were under-expressed in the bldG mutant when compared to the wt strain. For the bldA mutant, a total of 57 proteins were over-expressed and 1279 proteins were under-expressed when compared to the wt strain. A total of 224 proteins were over-expressed and 1240 were under-expressed in the bldA mutant when compared to the ΔbldG strain. Furthermore, 426, 222 and 227 proteins were expressed at similar levels between ΔbldG and wt, ΔbldA and wt, and ΔbldA and ΔbldG, respectively (Fig. 2a; Supplementary File 5). By sorting the differentially expressed proteins into functional groups (metabolism, cellular processes, regulation, energetics, translation) based on Pfam domains [29], it was found that the majority of proteins were differentially expressed in the bldG mutant when compared to the ΔbldA and wt strains (Fig. 2b). Of particular interest to this study was the identification of regulatory proteins that may be directly or indirectly regulated by BldG. This includes sigma factors and other transcriptional regulators that could potentially be involved in regulating secondary metabolism in S. clavuligerus. As shown in Fig. 3, 192 known and putative regulatory proteins showed statistically different expression levels among the three strains (Supplementary File 6). These 192 proteins along with others of interest based on predicted functions and the chromosomal location of their respective genes were chosen for further detailed analysis.

Pairwise analysis of differentially expressed proteins in the S. clavuligerus wt, ΔbldA and ΔbldG strains grown in SA medium. Differentially expressed proteins were analyzed per comparison (ΔbldG/wt, ΔbldA/wt and ΔbldA/ΔbldG) according to criteria described in the methods section. a Venn diagram showing number of proteins differentially expressed in the pairwise comparisons. The identities and details of proteins from each comparison and subgroup are listed in Supplementary File 5. b Number of differentially expressed proteins per comparison based on functional categories (metabolism, cellular processes, regulation, energetics and translation). Proteins deemed differentially expressed in more than one comparison were counted only once and were assigned to a comparison in the following order: ΔbldG/wt, ΔbldA/ΔbldG and ΔbldA/wt, respectively

Heat map showing the relative abundance of differentially expressed regulatory proteins in wt, ΔbldG, and ΔbldA strains of S. clavuligerus. Columns correspond to strains and replicates, whereas each row represents a protein included in the analysis. Proteins were grouped using hierarchical clustering with Euclidean distance and complete linkage. Heat map colors indicate row-wise Z scores. The Z score of a protein is the number of standard deviations away from the mean abundance of that protein across all samples. The dendrogram on the left indicates groups of similarly expressed proteins resulting from the hierarchical clustering analysis. Proteins with known regulatory functions from S. clavuligerus and other species are marked with a dash on the left panel. Details regarding the identities of the proteins and Z scores can be found in the supplementary section (Supplementary File 6)
Sigma factors and related proteins
A total of ten sigma factors, three ASFs, and one AASF were detected in the iTRAQ dataset (Table 1). Among these were the AASF encoded by bldG, two ASFs (Orf3/ApgA and PrsH/RshA) shown previously to interact with the BldG orthologue in S. coelicolor and S. griseus, and the sigma factor SigH, which in S. coelicolor and S. griseus is regulated by PrsH/RshA [86, 96]. As expected, BldG is under-expressed in both the S. clavuligerus bldG and bldA mutants as compared to the wt strain. However, the expression of Orf3/ApgA, PrsH/RshA and SigH was unaffected in the same pair-wise comparisons. Two additional sigma factors (BldN/AdsA and 490052605) were significantly under-expressed in the S. clavuligerus bldG mutant and were unaffected in the ΔbldA when compared to the wt strain, which agrees with previously published results on bldN expression in S. coelicolor bldA and bldG mutants [9]. A second sigma 70-family protein (490061073) was found to be significantly under-expressed in both the S. clavuligerus bldG and bldA mutants when compared to the wt strain. Additionally, four sigma factors were under-expressed in the ΔbldA when compared to the wt strain and were unaffected in the bldG mutant (Table 1). These include RpoE (known as SigR in S. coelicolor), which is a key regulator of the oxidative stress response [65]; HrdD, the function of which is yet unknown and is partially dependent on SigE for its own expression [17, 64]; a SigF homologue (490058906), which is required for proper sporulation in other Streptomyces spp. [98]; and HrdB, which is the principle sigma factor responsible for transcribing housekeeping genes in S. coelicolor [87]. In addition, two sigma 24-family proteins (497683136 and 490056752) were over-expressed in the bldG mutant and under-expressed in ΔbldA when compared to the wt strain (Table 1).
Transcriptional regulators
Several proteins that are known or may be involved in indirectly regulating CA production were detected in the dataset (Fig. 4). One AraC family protein (490055879/AdpA/BldH) is under-expressed in both the bldG and bldA mutants when compared to the wt strain. In addition, BldH/AdpA is under-expressed in ΔbldA when compared to ΔbldG, which can be explained by the presence of a TTA codon in bldH/adpA. The observed decrease in the expression of BldH/AdpA in the bldA mutant was not expected as it has a role in the positive regulation of ccaR and therefore indirectly controls CA biosynthesis [52]. A homologue of the S. coelicolor RNaseIII (490059733/AbsB), which is involved in regulating secondary metabolite production [74, 106], was under-expressed to similar extent in both ΔbldG and ΔbldA when compared to the wt strain. The expression of the S. clavuligerus BldD orthologue (490054092), which is known to repress the pre-mature expression of multiple genes involved in morphological differentiation and antibiotic biosynthesis in S. coelicolor [24, 25, 38], was unaffected in all comparisons (Supplementary Files 4–6). Finally, a protein identified as an AfsR-like regulator (497683347) was found to be similarly under-expressed in both ΔbldG and ΔbldA when compared to the wt strain. On closer examination, the protein was identified to be a partial entry for CcaR [2].

Regulatory cascade leading to clavulanic acid production and development in S. clavuligerus based on results from previously published reports [66] and the current study. Proteins and gene products detected in this study are indicated in purple color and are assigned a number, which was used to report their relative expression levels in the inset legend. Red and green arrows indicate repression or activation by the designated protein, respectively. In the inset legend G or A indicates the expression level of the respective protein assigned the specific number in the bldG or the bldA mutant as compared to the wt strain, with red indicating reduced expressed and green indicating over-expressed in the respective comparison
A putative AfsR-like protein (490057267) was under-expressed in ΔbldG when compared to the wt strain (Supplementary Files 4–6). The gene encoding this protein is located in a polyketide synthase (PKS)-associated gene cluster designated as SMC5, making it likely that this protein is a SARP [58]. A single LysR-family transcriptional regulator (497683951) was over-expressed in ΔbldG and under-expressed in ΔbldA when compared to the wt strain. Four IclR transcriptional regulators detected in the dataset were under-expressed in the bldG mutant when compared to the wt strain (Supplementary Files 4–6).
Six additional regulators were differentially expressed in the pairwise comparisons, which included proteins belonging to the AsnC-family (490055317 and 490056872), Fur-family (497681972), the PbsX-family (490050159/PcbR), NmrA-family (497683061) and DeoR-family (490052578) (Supplementary File 6). A single REX-family transcriptional regulator (490058535), which in other Streptomyces regulates genes encoding components of the respiratory chain [14], was found to be over-expressed in ΔbldG and under-expressed in ΔbldA when compared to the wt strain (Supplementary Files 4–6). Multiple GntR-family transcriptional regulators, which have various functions including gluconate repression, were also detected and were found to be differentially expressed in the comparisons (Supplementary Files 4–6). This included the auto regulatory sporulation specific transcriptional factor WhiH (490059845) [71], which was over-expressed in ΔbldA as compared to the ΔbldG strain.
Several TetR-family proteins were also identified and were found to be differentially expressed in the pairwise comparisons (Supplementary File 5). TetR-family members are involved in the transcriptional regulation of multidrug efflux, antibiotic biosynthesis pathways, osmotic stress response, catabolic pathways and differentiation processes [76]. One TetR-family protein (490060245) was under-expressed in the ΔbldG and was unaffected in the bldA mutant when compared to the wt strain. The gene encoding 490060245 (SCLAV_5246) is located downstream from the gene encoding RocG/GdhA, the deletion of which increases production of the immunosuppressant FK506 in S. tsukubaensis [37]. Two of the TetR-family proteins (497682848/AtrA and 490057306) were under-expressed in both ΔbldG and ΔbldA as compared to the wt strain. In S. coelicolor, AtrA regulates the transcription of actII-ORF4, the pathway-specific activator of actinorhodin biosynthesis [99]. The gene encoding the second TetR-family regulator (490057306: SCLAV_0504) is located upstream of genes involved in peptidoglycan synthesis (SCLAV_0506-0507). Two TetR-family regulators (490051466 and 497681723) were over-expressed in ΔbldG as compared to the wt strain and were unaffected in the bldA mutant. The protein designated as 490051466 is a homologue of AmfC, a mycelia formation protein that lacks any TetR-family regulatory domains. The gene encoding the second protein (497681723) is located downstream of putative genes encoding components of an ABC-type transport system (SCLAV1312-1313). An additional TetR-family regulator (497683062) was over-expressed in ΔbldG and under-expressed in the bldA mutant as compared to the wt strain. The gene encoding 497683062 (SCLAV_3466) is located in the opposite orientation next to genes encoding two putative serine/threonine protein kinases homologous to S. griseus AfsK (SCLAV_3467a and 3467b), as well as a gene encoding a hypothetical regulatory protein with a DUF397 domain (SCLAV_3468). Proteins containing the DUF397 domain have been previously implicated in regulating antibiotic production in S. coelicolor [28, 73].
A total of eight XRE-family transcriptional regulators, which control various and diverse metabolic functions, were also found to be differentially expressed in ΔbldG as compared to the wt strain (Table 2; Supplementary Files 4–6). The S. clavuligerus BldD orthologue (490054092), which is a pleiotropic XRE-family regulator controlling both development and antibiotic production in S. coelicolor, was not differentially expressed in ΔbldG or ΔbldA when compared to the wt strain.
Two component systems and serine/threonine kinases
PhoP (490059014), a response regulator known to be involved in the phosphate-mediated regulation of antibiotic biosynthesis [55], was found to be over-expressed in ΔbldG and was unaffected in ΔbldA when compared to the wt strain (Supplementary Files 4–6). Several of the proteins known to be under the control of PhoP were also observed to be differentially expressed, including PstB (490058978) and PhoU (490051157).
Several other response regulators were over-expressed in the bldG mutant, including the regulator of a vancomycin resistance system (490054741/VanR) [39] and 497682237, which is a putative orphan response regulator. Several other putative response regulator proteins belonging to the LuxR family were also differentially expressed in both ΔbldG and ΔbldA when compared to the wt strain (Supplementary File 4–6). Genes for all of these proteins were located in close proximity to genes encoding putative sensor kinases with the exception of 490056384, which is currently annotated in the database as a protein involved in differentiation.
A total of nine serine/threonine kinases were detected in the dataset (Supplementary File 4). The expression of the S. clavuligerus orthologue of the AfsK kinase (497683065) was unaffected in all comparisons. The AfsK/AfsR/AfsS system controls both differentiation and secondary metabolism and disruption of any of the encoding genes results in decreased antibiotic production [30, 34, 36, 50, 75]. The kinase designated as 497683146 was over-expressed in ΔbldG and under-expressed in ΔbldA when compared to the wt strain and is homologous to S. coelicolor PkaD, which is thought to be a positive regulator of actinorhodin production in that species [100]. Three other kinases known to be involved in development (490060020/AmfT/RamC, 497683145, and 497682612) were under-expressed in both ΔbldG and ΔbldA when compared to the wt strain. A kinase designated as 497683929 was over-expressed in ΔbldG and was unaffected in ΔbldA as compared to the wt strain. 497683929 is encoded by a gene (SCLAV_5481) located in a predicted non-ribosomal peptide associated gene cluster and does not seem to have any homologues in other sequenced Streptomyces species [58].
Identification and expression of proteins encoded by TTA codon containing genes
Analysis of the S. clavuligerus chromosome and plasmid (pSCL1, pSCL2, pSCL3 and pSCL4) nucleotide sequences identified a total of 243 TTA codon containing genes and 24 proteins encoded by them were present in the dataset, which included eight transcriptional regulators/DNA binding proteins (Table 3; Supplementary files 4, 5). Twenty proteins, including BldH/AdpA, were under-expressed in both ΔbldA and ΔbldG when compared to wt strain, but surprisingly none of the proteins were under-expressed solely in the bldA mutant. A total of 13 of the identified TTA codon containing genes in the data set have homologous counterparts in other Streptomyces species of which only five genes were found to retain the TTA codon in across all species (Table 3).
Discussion
The main goal of the described work was to identify proteome wide changes in the S. clavuligerus bldG and bldA mutants for comparison to each other and to the wt strain, focusing on proteins involved in regulatory pathways. Such analysis could lead to the elucidation of previously unknown regulatory pathways and mechanisms, which can provide avenues for manipulating and increasing CA production and secondary metabolism. In addition, regulatory elements found to be differentially expressed in the ΔbldG mutant when compared to the ΔbldA and wt strains could potentially be involved in the BldG-mediated regulatory cascade, which ultimately controls CA and cephamycin C production by regulating the expression of ccaR. The most relevant findings based on the described goals are discussed.
Additional sigma factor targets of BldG remain elusive
In Streptomyces, the expression of genes involved in both morphological differentiation and secondary metabolism require various sigma factors [9, 43, 85, 86, 103], but the sigma factor(s) responsible for the transcription of ccaR in S. clavuligerus is still unknown [11]. The BldG orthologue in S. coelicolor and S. griseus has been found to interact with several ASFs including Orf3/ApgA, PrsH/RshA (regulator of SigH) and RsfA (regulator of SigF) (Fig. 4) [60, 69, 86, 96]. The protein expression data obtained in the current study showed that BldG is under-expressed in both the S. clavuligerus bldG and bldA mutants when compared to the wt strain; however, the ASFs Orf3/ApgA and PrsH/RshA and the sigma factor SigH are not differentially expressed in any of the comparisons (Table 1; Fig. 4). This was surprising as bldG is necessary for sigH expression via the sigHp2 promoter in other Streptomyces species [86]. Therefore, the link between BldG and SigH in S. clavuligerus warrants further examination.
Further analysis was conducted to identify sigma-B and sigma-F family members in the dataset as these are known to be regulated by AASF/ASF protein pairs [6, 21]. The only sigma-B homologue detected in the dataset was HrdD, which was not differentially expressed in ΔbldG mutant and was under-expressed in ΔbldA when compared to the wt strain (Table 1). S. coelicolor hrdD mutants are not defective in development or secondary metabolism [17], making it unlikely that it is a target of BldG in S. clavuligerus. An S. clavuligerus SigF homologue (490058906) was also detected, which was under-expressed in both the bldG and bldA mutants when compared to the wt strain (Table 1). Previous studies have shown that SigF is required for proper sporulation S. coelicolor [98] and that BldG interacts with the SigF-specific ASF RsfA in that organism [60]. The reversible phosphorylation of BldG by RsfA is necessary for proper morphological development in S. coelicolor [10, 60], and both Orf3/ApgA and PrsH/RshA lack the necessary kinase domain required for this activity [69, 86]. It is likely that BldG has an additional sigma factor partner to interact with since sigH and sigF mutants of S. coelicolor exhibit a whi (white) phenotype in which aerial hyphae septation or maturation is affected, in contrast to the bld phenotype of the ΔbldG mutant [12, 85]. It is also likely that BldG has an additional ASF partner to interact with as in vivo phosphorylation of BldG was not affected in a previously analyzed S. coelicolor rsfA mutant [60].
Results also confirm a role for BldG in the regulation of BldN/AdsA, which is consistent with previously reported transcriptional studies in a S. coelicolor bldG mutant [9]. In the current study, BldN/AdsA is the most under-expressed sigma factor in ΔbldG as compared to the ΔbldA and the wt strains (Table 1). Both BldN/AdsA and the transcriptional regulator BldH/AdpA are required for the transcription of ssgA, which is necessary for spore formation in S. coelicolor [102, 107, 108], and BldH/AdpA also has a role in the positive regulation of ccaR expression in S. clavuligerus [52]. In S. coelicolor, BldN/AdsA is part of an operon that requires BldH/AdpA for its transcription [107], and BldH/AdpA is itself dependent on bldA for its translation due to the presence of a TTA codon in the encoding gene [95]. In the current study, BldH/AdpA was under-expressed in the S. clavuligerus bldA and bldG mutants when compared to the wt strain (Table 1). Since BldN/AdsA was not under-expressed in the bldA mutant when compared to the wt strain in this study, it appears that S. clavuligerus AdpA plays a role more similar to that of BldH/AdpA in S. coelicolor (BldH/AdpA not required for BldN/AdsA), rather than that of BldH/AdpA in S. griseus (BldH/AdpA required for BldN/AdsA) [95, 107]. This suggests that BldG somehow regulates BldN/AdsA expression independently of BldH/AdpA in S. clavuligerus. It is also possible that BldN/AdsA is the sigma factor responsible for the transcription of ccaR, which would be one of the first proteins to be identified to explain the link between BldG and ccaR expression (Fig. 4). Studies are currently underway to investigate the role of BldN/AdsA in the transcription of ccaR and other genes in S. clavuligerus.
A possible system of XRE-family transcriptional regulators and DUF397-domain containing proteins in the regulation of development and secondary metabolism
Several predicted DNA binding proteins were under-expressed in ΔbldG and showed varying expression patterns in ΔbldA when compared to the wt strain (Supplementary Files 4–6). Two of these predicted proteins (490051454 and 490054446) contain the HTH_XRE conserved domain found in the XRE-family of transcriptional regulators. The genes encoding 490051454 and 490054446 are clustered together with genes predicted to encode proteins containing the DUF397 domain, which has been previously implicated in the regulation of secondary metabolism [73]. A similar arrangement was observed for almost all of the genes encoding XRE-family proteins found to be under-expressed in the bldG mutant in the current study (497683074, 497682597, 490058590, 497681584, 490058041, 490055478, 490050197 and 490054541) (Table 2). There are 45 genes encoding putative DUF397-containing proteins in S. clavuligerus and only one (497683523) was present in the dataset, which was under-expressed in ΔbldG and was unaffected in ΔbldA when compared to the wt strain. In addition, 497683523 is 74 % similar to BldB, which has a known function in regulating development and secondary metabolism in S. coelicolor [73].
Previous studies have suggested that BldB and BldB-like proteins likely have interacting partners, the combinations of which somehow regulate cellular functions [22, 23]. Studies in S. coelicolor on an XRE-family regulator (WhiJ) and a small acidic protein of unknown function with a DUF397 domain (SCO4542) may provide clues as to how these proteins might regulate gene expression. Deletion of SCO4542, which is located immediately downstream of whiJ, results in a bald phenotype, but this phenotype is completely reversed in a SCO4542/whiJ double mutant [1]. This suggests that WhiJ represses development and the SCO4542 product somehow counters its ability to do so. Therefore, it is possible that the function of the DUF397 domain containing proteins is to interact antagonistically with XRE-family proteins [57]. Based on the arrangement of the XRE-family/DUF397 containing protein encoding genes in S. clavuligerus and the proteomics data (Table 2), it is possible that these systems have some role in regulating secondary metabolism. In addition, these genes could themselves be under the indirect control of BldG, making them good candidates for future manipulation studies to elucidate their roles in CA production.
Possible role for BldD/BtdA in regulating CA production in S. clavuligerus
The most under-expressed transcriptional regulator in the S. clavuligerus bldG mutant (slightly over-expressed in ΔbldA) as compared to the wt strain is BdtA (Supplementary Files 4–6). The btdA gene encodes a DNA-binding protein that contains a MerR-family domain, and was originally thought to have a role in development in S. coelicolor [25]. Later studies showed that a S. coelicolor bdtA mutant did not have a distinctive phenotype [20, 38]. BldC is the only BdtA homologue in S. coelicolor and it is required for both differentiation and secondary metabolite biosynthesis [38]. The bdtA gene is negatively regulated by the pleiotropic transcriptional regulator BldD, which is known to repress the pre-mature expression of multiple genes involved in development, including the genes encoding the sigma factors WhiG and BldN/AdsA, as well as bldC (Fig. 4) [20]. Both BldD and BldC were detected in the dataset, with BldD not being differentially expressed in any of the comparisons and BldC being under-expressed in only the bldA mutant when compared to the wt strain (Supplementary Files 4–6).
At this point, it is not possible to determine whether BdtA is critical for either development or secondary metabolite biosynthesis in S. clavuligerus. However, the current expression data suggests an indirect role of BldG in the regulation of bdtA. In S. coelicolor, BldG and BldD are already known to indirectly or directly regulate expression of the same gene product in at least one case, namely SigH. Transcription from the SigH dependent promoter sigHp2 is abolished in a bldG mutant [86] and is activated in a bldD mutant [45]. Therefore, results from the current study are in good agreement with those from previous reports and work is currently underway to investigate a possible link between BdtA and CA production in S. clavuligerus.
BldG and phosphate regulation of secondary metabolism
The PhoP response regulator was found to be over-expressed in ΔbldG and was unaffected in ΔbldA when compared to the wt strain (Supplementary Files 4–6). The PhoR–PhoP TCS is involved in the phosphate-mediated regulation of secondary metabolism in Streptomyces [55]. Deletion of the system results in varying phenotypes depending on the host, suggesting that phosphate-mediated regulation of secondary metabolism is stricter in some species [32, 89]. For example, PhoP deletion mutants over-produce pigmented secondary metabolites in S. lividans [89], but no such increase is observed in S. coelicolor [84]. Additionally, proteins involved in phosphate transport that are positively regulated by PhoP under conditions of phosphate limitation were also found to be differentially expressed [32, 80], including the pathway-specific activator PhoU (unaffected in ΔbldG and under-expressed in ΔbldA when compared to the wt strain) and PstB (under-expressed in ΔbldG and unaffected in ΔbldA when compared to the wt strain) (Supplementary Files 4–6). PhoU plays a role in the regulation of phosphate uptake and its expression has previously been found to be induced when PhoP is phosphorylated [90], and PstB is a PhoP-dependent member of the phosphate transport system [79]. The pho regulon has been characterized in S. clavuligerus (Fig. 4) and CA production is negatively regulated by phosphate [49]. It has also been suggested that PhoP somehow indirectly regulates CA biosynthesis since DNA binding sites for the protein were not identified in the CA biosynthetic gene cluster [80]. Therefore, the over-expression of PhoP in the bldG mutant could help explain the lack of CA production in this strain, although the link between BldG and PhoP is unclear at this time.
TTA dependence is re-evaluated in S. clavuligerus
In Streptomyces species, the bldA gene specifies the only known t-RNA capable of translating rare leucine TTA codons [72, 105], and S. coelicolor bldA mutants are blocked in both development and secondary metabolism [35, 48, 59]. It is also known that bldH/adpA encodes the pleiotropic transcriptional regulator responsible for the ΔbldA phenotype [77], and several other TTA-containing genes are required for the biosynthesis of the blue-pigmented secondary metabolite actinorhodin in S. coelicolor [27, 95]. In S. clavuligerus, bldA is only essential for development and not for secondary metabolism, which was surprising as ccaR contains a TTA codon [97].
It was suggested that certain TTA codons such as those present in ccaR are effectively mistranslated based on their location within the gene and the nucleotides flanking them [97]. The presence of rare codons within the first 25–50 codons has been shown to have a more severe effect on translation as compared to their presence towards 3′ end of the transcript [19]. This is unlikely to be the case for ccaR, which has a TTA codon at position 38 [97]. It was suggested that the identity of the nucleotides flanking the TTA codon in ccaR may allow it to be mistranslated, based on other systems involving frame shifting in yeasts and viruses [4, 15, 97]. According to the current hypothesis, a bldA-dependent TTA codon is immediately followed by a C or a T at the 3′ position, whereas a codon capable of being mistranslated such as the one in ccaR has a G at this position [97]. To date, ccaR was the only known bldA-independent TTA codon containing gene. Therefore, it was hypothesized that bldA-independent genes would always have a G or potentially an A at the following 3′ position of the TTA codon [97].
A total of 24 out of 243 predicted proteins (~10 %) encoded by TTA containing genes were present in the dataset (Table 3; Supplementary Files 4–5), which is a much lower percentage as compared to the total proteome detected. This is likely in part due to the fact that TTA codons only occur in genes involved in secondary metabolism and morphological differentiation, and therefore were not expressed under the growth conditions used [51]. AdpA/BldH appears to follow the conventional pattern of expression in that it is under-expressed in the bldA mutant and contains a C immediately following the TTA codon [95]; however, the model does not seem to fit well in other cases. Four proteins found to be most under-expressed in ΔbldA mutant when compared to ΔbldG and the wt strain are also encoded by genes containing a G or an A following the TTA codon (497681088, 490057169, 497685265, 490058041) (Table 3), suggesting that the presence of these nucleotides does not necessarily allow for effective mistranslation. The expression of three proteins (490060397, 490058513 and 497685280) encoded by genes with a C at the immediately 3′ end of the TTA codon was either unaffected or they were over-expressed in the bldA mutant when compared to the ΔbldG and wt strains, suggesting that all genes with Cs following the TTA codon are not bldA-dependent. Therefore, results from the current study do not support the nucleotide based mistranslation hypothesis described above [97], which is consistent with the slightly different biological programming followed in S. clavuligerus as compared to other model Streptomyces spp.
In conclusion, a total of 2442 proteins corresponding to 33.5 % of the S. clavuligerus proteome were detected from cellular lysates in the current study. There are only three previously published reports on the analysis of Streptomyces proteomes using iTRAQ in the literature, which were able to detect up to 15 % of the predicted proteome of S. coelicolor [40, 53, 62]. Many proteins involved in regulation were found to be differentially expressed in the comparisons (Figs. 3, 4). The genes for some of the differentially expressed proteins identified are good candidates for future manipulation studies for increasing CA production in S. clavuligerus. In addition, the analysis of proteins encoded by TTA codon containing genes suggests that the role of bldA and TTA dependence in S. clavuligerus needs to be reconsidered. To the best of our knowledge, the described study represents the most in depth proteomic cataloging and analysis in S. clavuligerus to date. The reported proteomics data will serve in the future as an useful tool to direct research into aspects of secondary metabolism, as well as into aspects of development and primary metabolism in S. clavuligerus and other industrially important Streptomyces species.
Abbreviations
- AASF:
-
Anti–anti-sigma factor
- ASF:
-
Anti-sigma factor
- CA:
-
Clavulanic acid
- CDD:
-
Conserved domain database
- dcGO:
-
Database of domain-centric ontologies
- FDR:
-
False discovery rate
- FT:
-
Fourier transformed
- HCD:
-
Higher-energy collisional dissociation
- HPLC:
-
High performance liquid chromatography
- iTRAQ:
-
Isobaric tags for relative and absolute quantitation
- ISP:
-
International Streptomyces project
- LC:
-
Liquid chromatography
- MYM:
-
Maltose-yeast extract-malt extract
- MMTS:
-
Methyl methanethiosulfonate
- MS/MS:
-
Tandem mass spectrometer/spectrometry
- SA:
-
Starch asparagine
- SARP:
-
Streptomyces antibiotic regulatory protein
- SDS:
-
Sodium dodecyl sulfate
- TCEP:
-
Tris(2-carboxyethyl)phosphine hydrochloride
- TCS:
-
Two component system
- TEAB:
-
Triethylammonium bicarbonate
- TSBS:
-
Trypticase soy broth with starch
- wt:
-
Wild type
References
Ainsa JA, Bird N, Ryding NJ, Findlay KC, Chater KF (2010) The complex whiJ locus mediates environmentally sensitive repression of development of Streptomyces coelicolor A3(2). Antonie Van Leeuwenhoek 98:225–236
Alexander DC, Jensen SE (1998) Investigation of the Streptomyces clavuligerus cephamycin C gene cluster and its regulation by the CcaR protein. J Bacteriol 180:4068–4079
Alvarez-Alvarez R, Rodriguez-Garcia A, Santamarta I, Perez-Redondo R, Prieto-Dominguez A, Martinez-Burgo Y, Liras P (2014) Transcriptomic analysis of Streptomyces clavuligerus Delta-ccaR:tsr: effects of the cephamycin C-clavulanic acid cluster regulator CcaR on global regulation. Microb Biotechnol 7:221–231
Belcourt MF, Farabaugh PJ (1990) Ribosomal frameshifting in the yeast retrotransposon Ty: tRNAs induce slippage on a 7 nucleotide minimal site. Cell 62:339–352
Benjamini YaH Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 57:289–300
Benson AK, Haldenwang WG (1993) Bacillus subtilis sigma B is regulated by a binding protein (RsbW) that blocks its association with core RNA polymerase. Proc Natl Acad Sci USA 90:2330–2334
Bentley PH, Berry PD, Brooks G, Gilpin ML, Hunt E, Zomaya II (1977) Total synthesis of (±)-clavulanic acid. J Chem Soc Chem Commun 748–749
Bibb MJ (2005) Regulation of secondary metabolism in streptomycetes. Curr Opin Microbiol 8:208–215
Bibb MJ, Molle V, Buttner MJ (2000) Sigma(BldN), an extracytoplasmic function RNA polymerase sigma factor required for aerial mycelium formation in Streptomyces coelicolor A3(2). J Bacteriol 182:4606–4616
Bignell DR, Lau LH, Colvin KR, Leskiw BK (2003) The putative anti–anti-sigma factor BldG is post-translationally modified by phosphorylation in Streptomyces coelicolor. FEMS Microbiol Lett 225:93–99
Bignell DR, Tahlan K, Colvin KR, Jensen SE, Leskiw BK (2005) Expression of ccaR, encoding the positive activator of cephamycin C and clavulanic acid production in Streptomyces clavuligerus, is dependent on bldG. Antimicrob Agents Chemother 49:1529–1541
Bignell DR, Warawa JL, Strap JL, Chater KF, Leskiw BK (2000) Study of the bldG locus suggests that an anti–anti-sigma factor and an anti-sigma factor may be involved in Streptomyces coelicolor antibiotic production and sporulation. Microbiology 146(Pt 9):2161–2173
Boehm AM, Putz S, Altenhofer D, Sickmann A, Falk M (2007) Precise protein quantification based on peptide quantification using iTRAQ. BMC Bioinform 8:214. doi:10.1186/1471-2105-8-214
Brekasis D, Paget MS (2003) A novel sensor of NADH/NAD + redox poise in Streptomyces coelicolor A3(2). EMBO J 22:4856–4865
Brierley I, Jenner AJ, Inglis SC (1992) Mutational analysis of the “slippery-sequence” component of a coronavirus ribosomal frameshifting signal. J Mol Biol 227:463–479
Brown D, Evans JR, Fletton RA (1979) Structures of three novel beta-lactams isolated from Streptomyces clavuligerus. Chem Commun 6:282–283
Buttner MJ, Chater KF, Bibb MJ (1990) Cloning, disruption, and transcriptional analysis of three RNA polymerase sigma factor genes of Streptomyces coelicolor A3(2). J Bacteriol 172:3367–3378
Casado-Vela J, Martinez-Esteso MJ, Rodriguez E, Borras E, Elortza F, Bru-Martinez R (2010) iTRAQ-based quantitative analysis of protein mixtures with large fold change and dynamic range. Proteomics 10:343–347
Chen GF, Inouye M (1990) Suppression of the negative effect of minor arginine codons on gene expression; preferential usage of minor codons within the first 25 codons of the Escherichia coli genes. Nucleic Acids Res 18:1465–1473
den Hengst CD, Tran NT, Bibb MJ, Chandra G, Leskiw BK, Buttner MJ (2010) Genes essential for morphological development and antibiotic production in Streptomyces coelicolor are targets of BldD during vegetative growth. Mol Microbiol 78:361–379
Duncan L, Losick R (1993) SpoIIAB is an anti-sigma factor that binds to and inhibits transcription by regulatory protein sigma F from Bacillus subtilis. Proc Natl Acad Sci USA 90:2325–2329
Eccleston M, Ali RA, Seyler R, Westpheling J, Nodwell J (2002) Structural and genetic analysis of the BldB protein of Streptomyces coelicolor. J Bacteriol 184:4270–4276
Eccleston M, Willems A, Beveridge A, Nodwell JR (2006) Critical residues and novel effects of overexpression of the Streptomyces coelicolor developmental protein BldB: evidence for a critical interacting partner. J Bacteriol 188:8189–8195
Elliot M, Damji F, Passantino R, Chater K, Leskiw B (1998) The bldD gene of Streptomyces coelicolor A3(2): a regulatory gene involved in morphogenesis and antibiotic production. J Bacteriol 180:1549–1555
Elliot MA, Bibb MJ, Buttner MJ, Leskiw BK (2001) BldD is a direct regulator of key developmental genes in Streptomyces coelicolor A3(2). Mol Microbiol 40:257–269
Fang H, Gough J (2013) DcGO: database of domain-centric ontologies on functions, phenotypes, diseases and more. Nucleic Acids Res 41:D536–D544
Fernandez-Moreno MA, Caballero JL, Hopwood DA, Malpartida F (1991) The act cluster contains regulatory and antibiotic export genes, direct targets for translational control by the bldA tRNA gene of Streptomyces. Cell 66:769–780
Fernandez-Moreno MA, Martin-Triana AJ, Martinez E, Niemi J, Kieser HM, Hopwood DA, Malpartida F (1992) abaA, a new pleiotropic regulatory locus for antibiotic production in Streptomyces coelicolor. J Bacteriol 174:2958–2967
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222–D230
Floriano B, Bibb M (1996) afsR is a pleiotropic but conditionally required regulatory gene for antibiotic production in Streptomyces coelicolor A3(2). Mol Microbiol 21:385–396
Foulstone M, Reading C (1982) Assay of amoxicillin and clavulanic acid, the components of Augmentin, in biological fluids with high-performance liquid chromatography. Antimicrob Agents Chemother 22:753–762
Ghorbel S, Kormanec J, Artus A, Virolle MJ (2006) Transcriptional studies and regulatory interactions between the phoR–phoP operon and the phoU, mtpA, and ppk genes of Streptomyces lividans TK24. J Bacteriol 188:677–686
Guthrie EP, Chater KF (1990) The level of a transcript required for production of a Streptomyces coelicolor antibiotic is conditionally dependent on a tRNA gene. J Bacteriol 172:6189–6193
Hempel AM, Cantlay S, Molle V, Wang SB, Naldrett MJ, Parker JL, Richards DM, Jung YG, Buttner MJ, Flardh K (2012) The Ser/Thr protein kinase AfsK regulates polar growth and hyphal branching in the filamentous bacteria Streptomyces. Proc Natl Acad Sci USA 109:E2371–E2379
Hopwood DA (1967) Genetic analysis and genome structure in Streptomyces coelicolor. Bacteriol Rev 31:373–403
Horinouchi S, Hara O, Beppu T (1983) Cloning of a pleiotropic gene that positively controls biosynthesis of A-factor, actinorhodin, and prodigiosin in Streptomyces coelicolor A3(2) and Streptomyces lividans. J Bacteriol 155:1238–1248
Huang D, Xia M, Li S, Wen J, Jia X (2013) Enhancement of FK506 production by engineering secondary pathways of Streptomyces tsukubaensis and exogenous feeding strategies. J Ind Microbiol Biotechnol 40:1023–1037
Hunt AC, Servin-Gonzalez L, Kelemen GH, Buttner MJ (2005) The bldC developmental locus of Streptomyces coelicolor encodes a member of a family of small DNA-binding proteins related to the DNA-binding domains of the MerR family. J Bacteriol 187:716–728
Hutchings MI, Hong HJ, Buttner MJ (2006) The vancomycin resistance VanRS two-component signal transduction system of Streptomyces coelicolor. Mol Microbiol 59:923–935
Jayapal KP, Philp RJ, Kok YJ, Yap MG, Sherman DH, Griffin TJ, Hu WS (2008) Uncovering genes with divergent mRNA-protein dynamics in Streptomyces coelicolor. PLoS One 3:e2097. doi:10.1371/journal.pone.0002097
Jensen SE (2012) Biosynthesis of clavam metabolites. J Ind Microbiol Biotechnol 39:1407–1419
Jensen SE, Paradkar AS, Mosher RH, Anders C, Beatty PH, Brumlik MJ, Griffin A, Barton B (2004) Five additional genes are involved in clavulanic acid biosynthesis in Streptomyces clavuligerus. Antimicrob Agents Chemother 48:192–202
Kelemen GH, Brown GL, Kormanec J, Potuckova L, Chater KF, Buttner MJ (1996) The positions of the sigma-factor genes, whiG and sigF, in the hierarchy controlling the development of spore chains in the aerial hyphae of Streptomyces coelicolor A3(2). Mol Microbiol 21:593–603
Kelemen GH, Buttner MJ (1998) Initiation of aerial mycelium formation in Streptomyces. Curr Opin Microbiol 1:656–662
Kelemen GH, Viollier PH, Tenor J, Marri L, Buttner MJ, Thompson CJ (2001) A connection between stress and development in the multicellular prokaryote Streptomyces coelicolor A3(2). Mol Microbiol 40:804–814
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. John Innes Centre, Norwich
Kwak J, Kendrick KE (1996) Bald mutants of Streptomyces griseus that prematurely undergo key events of sporulation. J Bacteriol 178:4643–4650
Lawlor EJ, Baylis HA, Chater KF (1987) Pleiotropic morphological and antibiotic deficiencies result from mutations in a gene encoding a tRNA-like product in Streptomyces coelicolor A3(2). Genes Dev 1:1305–1310
Lebrihi A, Germain D, Lefebvre G (1987) Phosphate repression of cephamycin and clavulanic acid production by Streptomyces clavuligerus. Appl Microbiol Biotechnol 26:130–135
Lee Y, Kim K, Suh JW, Rhee S, Lim Y (2007) Binding study of AfsK, a Ser/Thr kinase from Streptomyces coelicolor A3(2) and S-adenosyl-l-methionine. FEMS Microbiol Lett 266:236–240
Leskiw BK, Lawlor EJ, Fernandez-Abalos JM, Chater KF (1991) TTA codons in some genes prevent their expression in a class of developmental, antibiotic-negative, Streptomyces mutants. Proc Natl Acad Sci USA 88:2461–2465
Lopez-Garcia MT, Santamarta I, Liras P (2010) Morphological differentiation and clavulanic acid formation are affected in a Streptomyces clavuligerus adpA-deleted mutant. Microbiology 156:2354–2365
Manteca A, Sanchez J, Jung HR, Schwammle V, Jensen ON (2010) Quantitative proteomics analysis of Streptomyces coelicolor development demonstrates that onset of secondary metabolism coincides with hypha differentiation. Mol Cell Proteom 9:1423–1436
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39:D225–D229
Martin JF (2004) Phosphate control of the biosynthesis of antibiotics and other secondary metabolites is mediated by the PhoR–PhoP system: an unfinished story. J Bacteriol 186:5197–5201
Martinez-Burgo Y, Alvarez-Alvarez R, Rodriguez-Garcia A, Liras P (2015) The pathway-specific regulator ClaR of Streptomyces clavuligerus has a global effect on the expression of genes for secondary metabolism and differentiation. Appl Environ Microbiol 81:6637–6648
McCormick JR, Flardh K (2012) Signals and regulators that govern Streptomyces development. FEMS Microbiol Rev 36:206–231
Medema MH, Trefzer A, Kovalchuk A, van den Berg M, Muller U, Heijne W, Wu L, Alam MT, Ronning CM, Nierman WC, Bovenberg RA, Breitling R, Takano E (2010) The sequence of a 1.8-mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol Evol 2:212–224
Merrick MJ (1976) A morphological and genetic mapping study of bald colony mutants of Streptomyces coelicolor. J Gen Microbiol 96:299–315
Mingyar E, Sevcikova B, Rezuchova B, Homerova D, Novakova R, Kormanec J (2014) The sigma(F)-specific anti-sigma factor RsfA is one of the protein kinases that phosphorylates the pleiotropic anti–anti-sigma factor BldG in Streptomyces coelicolor A3(2). Gene 538:280–287
Molle V, Buttner MJ (2000) Different alleles of the response regulator gene bldM arrest Streptomyces coelicolor development at distinct stages. Mol Microbiol 36:1265–1278
Navone L, Casati P, Licona-Cassani C, Marcellin E, Nielsen LK, Rodriguez E, Gramajo H (2014) Allantoin catabolism influences the production of antibiotics in Streptomyces coelicolor. Appl Microbiol Biotechnol 98:351–360
Nguyen KT, Tenor J, Stettler H, Nguyen LT, Nguyen LD, Thompson CJ (2003) Colonial differentiation in Streptomyces coelicolor depends on translation of a specific codon within the adpA gene. J Bacteriol 185:7291–7296
Paget MS, Chamberlin L, Atrih A, Foster SJ, Buttner MJ (1999) Evidence that the extracytoplasmic function sigma factor sigma E is required for normal cell wall structure in Streptomyces coelicolor A3(2). J Bacteriol 181:204–211
Paget MS, Kang JG, Roe JH, Buttner MJ (1998) Sigma R, an RNA polymerase sigma factor that modulates expression of the thioredoxin system in response to oxidative stress in Streptomyces coelicolor A3(2). EMBO J 17:5776–5782
Paradkar A (2013) Clavulanic acid production by Streptomyces clavuligerus: biogenesis, regulation and strain improvement. J Antibiot (Tokyo) 66:411–420
Paradkar AS, Aidoo KA, Jensen SE (1998) A pathway-specific transcriptional activator regulates late steps of clavulanic acid biosynthesis in Streptomyces clavuligerus. Mol Microbiol 27:831–843
Paradkar AS, Jensen SE (1995) Functional analysis of the gene encoding the clavaminate synthase 2 isoenzyme involved in clavulanic acid biosynthesis in Streptomyces clavuligerus. J Bacteriol 177:1307–1314
Parashar A, Colvin KR, Bignell DR, Leskiw BK (2009) BldG and SCO3548 interact antagonistically to control key developmental processes in Streptomyces coelicolor. J Bacteriol 191:2541–2550
Perez-Llarena FJ, Liras P, Rodriguez-Garcia A, Martin JF (1997) A regulatory gene (ccaR) required for cephamycin and clavulanic acid production in Streptomyces clavuligerus: amplification results in overproduction of both beta-lactam compounds. J Bacteriol 179:2053–2059
Persson J, Chater KF, Flardh K (2013) Molecular and cytological analysis of the expression of Streptomyces sporulation regulatory gene whiH. FEMS Microbiol Lett 341:96–105
Piret JM, Chater KF (1985) Phage-mediated cloning of bldA, a region involved in Streptomyces coelicolor morphological development, and its analysis by genetic complementation. J Bacteriol 163:965–972
Pope MK, Green B, Westpheling J (1998) The bldB gene encodes a small protein required for morphogenesis, antibiotic production, and catabolite control in Streptomyces coelicolor. J Bacteriol 180:1556–1562
Price B, Adamidis T, Kong R, Champness W (1999) A Streptomyces coelicolor antibiotic regulatory gene, absB, encodes an RNase III homolog. J Bacteriol 181:6142–6151
Rajkarnikar A, Kwon HJ, Ryu YW, Suh JW (2006) Catalytic domain of AfsKav modulates both secondary metabolism and morphologic differentiation in Streptomyces avermitilis ATCC 31272. Curr Microbiol 53:204–208
Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang X, Gallegos MT, Brennan R, Tobes R (2005) The TetR family of transcriptional repressors. Microbiol Mol Biol Rev 69:326–356
Reading C, Cole M (1977) Clavulanic acid: a beta-lactamase-inhibiting beta-lactam from Streptomyces clavuligerus. Antimicrob Agents Chemother 11:852–857
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. doi:10.1093/nar/gkv007
Rodriguez-Garcia A, Barreiro C, Santos-Beneit F, Sola-Landa A, Martin JF (2007) Genome-wide transcriptomic and proteomic analysis of the primary response to phosphate limitation in Streptomyces coelicolor M145 and in a Delta-phoP mutant. Proteomics 7:2410–2429
Salehghamari E, Hamedi J, Elahi E, Sepehrizadeh Z, Sadeghi M, Muth G (2012) Prediction of the pho regulon in Streptomyces clavuligerus DSM 738. New Microbiol 35:447–457
Sanchez L, Brana AF (1996) Cell density influences antibiotic biosynthesis in Streptomyces clavuligerus. Microbiology 142(Pt 5):1209–1220
Santamarta I, Lopez-Garcia MT, Kurt A, Nardiz N, Alvarez-Alvarez R, Perez-Redondo R, Martin JF, Liras P (2011) Characterization of DNA-binding sequences for CcaR in the cephamycin-clavulanic acid supercluster of Streptomyces clavuligerus. Mol Microbiol 81:968–981
Santamarta I, Rodriguez-Garcia A, Perez-Redondo R, Martin JF, Liras P (2002) CcaR is an autoregulatory protein that binds to the ccaR and cefD-cmcI promoters of the cephamycin C-clavulanic acid cluster in Streptomyces clavuligerus. J Bacteriol 184:3106–3113
Santos-Beneit F, Rodriguez-Garcia A, Sola-Landa A, Martin JF (2009) Cross-talk between two global regulators in Streptomyces: PhoP and AfsR interact in the control of afsS, pstS and phoRP transcription. Mol Microbiol 72:53–68
Sevcikova B, Benada O, Kofronova O, Kormanec J (2001) Stress-response sigma factor (sigma H) is essential for morphological differentiation of Streptomyces coelicolor A3(2). Arch Microbiol 177:98–106
Sevcikova B, Rezuchova B, Homerova D, Kormanec J (2010) The anti–anti-sigma factor BldG is involved in activation of the stress response sigma factor (sigma H) in Streptomyces coelicolor A3(2). J Bacteriol 192:5674–5681
Shiina T, Tanaka K, Takahashi H (1991) Sequence of hrdB, an essential gene encoding sigma-like transcription factor of Streptomyces coelicolor A3(2): homology to principal sigma factors. Gene 107:145–148
Smyth GK (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3(article 3). doi:10.2202/1544-6115.1027
Sola-Landa A, Moura RS, Martin JF (2003) The two-component PhoR–PhoP system controls both primary metabolism and secondary metabolite biosynthesis in Streptomyces lividans. Proc Natl Acad Sci USA 100:6133–6138
Sola-Landa A, Rodriguez-Garcia A, Franco-Dominguez E, Martin JF (2005) Binding of PhoP to promoters of phosphate-regulated genes in Streptomyces coelicolor: identification of PHO boxes. Mol Microbiol 56:1373–1385
Song JY, Jensen SE, Lee KJ (2010) Clavulanic acid biosynthesis and genetic manipulation for its overproduction. Appl Microbiol Biotechnol 88:659–669
Song JY, Jeong H, Yu DS, Fischbach MA, Park HS, Kim JJ, Seo JS, Jensen SE, Oh TK, Lee KJ, Kim JF (2010) Draft genome sequence of Streptomyces clavuligerus NRRL 3585, a producer of diverse secondary metabolites. J Bacteriol 192:6317–6318
Tahlan K, Anders C, Jensen SE (2004) The paralogous pairs of genes involved in clavulanic acid and clavam metabolite biosynthesis are differently regulated in Streptomyces clavuligerus. J Bacteriol 186:6286–6297
Tahlan K, Park HU, Jensen SE (2004) Three unlinked gene clusters are involved in clavam metabolite biosynthesis in Streptomyces clavuligerus. Can J Microbiol 50:803–810
Takano E, Tao M, Long F, Bibb MJ, Wang L, Li W, Buttner MJ, Bibb MJ, Deng ZX, Chater KF (2003) A rare leucine codon in adpA is implicated in the morphological defect of bldA mutants of Streptomyces coelicolor. Mol Microbiol 50:475–486
Takano H, Fujimoto M, Urano H, Beppu T, Ueda K (2011) Cross-interaction of anti-sigmaH factor RshA with BldG, an anti-sigma factor antagonist in Streptomyces griseus. FEMS Microbiol Lett 314:158–163. doi:10.1111/j.1574-6968.2010.02155.x
Trepanier NK, Jensen SE, Alexander DC, Leskiw BK (2002) The positive activator of cephamycin C and clavulanic acid production in Streptomyces clavuligerus is mistranslated in a bldA mutant. Microbiology 148:643–656
Tzanis A, Dalton KA, Hesketh A, den Hengst CD, Buttner MJ, Thibessard A, Kelemen GH (2014) A sporulation-specific, sigF-dependent protein, SspA, affects septum positioning in Streptomyces coelicolor. Mol Microbiol 91:363–380
Uguru GC, Stephens KE, Stead JA, Towle JE, Baumberg S, McDowall KJ (2005) Transcriptional activation of the pathway-specific regulator of the actinorhodin biosynthetic genes in Streptomyces coelicolor. Mol Microbiol 58:131–150
Urabe H, Aoyagi N, Ogawara H, Motojima K (2008) Expression and characterization of the Streptomyces coelicolor serine/threonine protein kinase PkaD. Biosci Biotechnol Biochem 72:778–785
van Wezel GP, McDowall KJ (2011) The regulation of the secondary metabolism of Streptomyces: new links and experimental advances. Nat Prod Rep 28:1311–1333
van Wezel GP, van der Meulen J, Kawamoto S, Luiten RG, Koerten HK, Kraal B (2000) ssgA is essential for sporulation of Streptomyces coelicolor A3(2) and affects hyphal development by stimulating septum formation. J Bacteriol 182:5653–5662
Viollier PH, Kelemen GH, Dale GE, Nguyen KT, Buttner MJ, Thompson CJ (2003) Specialized osmotic stress response systems involve multiple SigB-like sigma factors in Streptomyces coelicolor. Mol Microbiol 47:699–714
Ward JM, Hodgson JE (1993) The biosynthetic genes for clavulanic acid and cephamycin production occur as a ‘super-cluster’ in three Streptomyces. FEMS Microbiol Lett 110:239–242
Wright F, Bibb MJ (1992) Codon usage in the G + C-rich Streptomyces genome. Gene 113:55–65
Xu W, Huang J, Cohen SN (2008) Autoregulation of AbsB (RNase III) expression in Streptomyces coelicolor by endoribonucleolytic cleavage of absB operon transcripts. J Bacteriol 190:5526–5530
Yamazaki H, Ohnishi Y, Horinouchi S (2000) An A-factor-dependent extracytoplasmic function sigma factor (AdsA) that is essential for morphological development in Streptomyces griseus. J Bacteriol 182:4596–4605
Yamazaki H, Ohnishi Y, Horinouchi S (2003) Transcriptional switch on of ssgA by A-factor, which is essential for spore septum formation in Streptomyces griseus. J Bacteriol 185:1273–1283
Zaburannyy N, Ostash B, Fedorenko V (2009) TTA Lynx: a web-based service for analysis of actinomycete genes containing rare TTA codon. Bioinformatics 25:2432–2433
Acknowledgments
The described work was funded by start-up and operating grants from the Research and Development Corporation of Newfoundland and Labrador (RDC: 5404.1218.102) and the Natural Science and Engineering Research Council of Canada (NSERC: 386417-2010) to KT, respectively. Equipment used in the study was purchased with grants from the Canadian Foundation for Innovation (CFI), RDC and NSERC to KT and DRDB. NLF and MAM also received support from the Memorial University of Newfoundland to pursue their programs of study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
Cite this article
Ferguson, N.L., Peña-Castillo, L., Moore, M.A. et al. Proteomics analysis of global regulatory cascades involved in clavulanic acid production and morphological development in Streptomyces clavuligerus . J Ind Microbiol Biotechnol 43, 537–555 (2016). https://doi.org/10.1007/s10295-016-1733-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-016-1733-y