
Abstract
Here, we attempted to elevate poly-gamma-glutamic acid (γ-PGA) production by modifying genes involved in glutamate metabolism in Bacillus amyloliquefaciens LL3. Products of rocR, rocG and gudB facilitate the conversion from glutamate to 2-oxoglutarate in Bacillus subtillis. The gene odhA is responsible for the synthesis of a component of the 2-oxoglutarate dehydrogenase complex that catalyzes the oxidative decarboxylation of 2-oxoglutarate to succinyl coenzyme A. In-frame deletions of these four genes were performed. In shake flask experiments the gudB/rocG double mutant presented enhanced production of γ-PGA, a 38 % increase compared with wild type. When fermented in a 5-L fermenter with pH control, the γ-PGA yield of the rocR mutant was increased to 5.83 g/L from 4.55 g/L for shake flask experiments. The gudB/rocG double mutant produced 5.68 g/L γ-PGA compared with that of 4.03 g/L for the wild type, a 40 % increase. Those results indicated the possibility of improving γ-PGA production by modifying glutamate metabolism, and identified potential genetic targets to improve γ-PGA production.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Poly-γ-glutamic acid (γ-PGA) is a naturally occurring polymer that has attracted extensive investigations due to its outstanding qualities, such as water solubility, biocompatibility and degradability [19]. Composed of L- and D-glutamic acids linked by γ-amide linkages, γ-PGA has a wide range of applications ranging from hydrogels, flocculants, drug delivery, cosmetics to feed additives [2–4, 17, 22]. Regardless of its valuable characteristics, the practical use of γ-PGA is hindered mostly by its high cost compared with conventional materials in current use. Improving microbial γ-PGA production, especially using molecular biology techniques, is thus an important goal in both scientific and industrial fields. However, only very limited information is available regarding the genetic elements that can be targeted to improve existing γ-PGA producers. To the best of our knowledge, those elements include a gene responsible for the synthesis of exopolysaccharides, genes encoding γ-PGA degradation enzymes and the vgb gene coding for hemoglobin [12, 15, 18, 21].
We previously reported that the deletion of RocR, a transcriptional regulator in glutamate metabolism, may have contributed to the increase of γ-PGA production in Bacillus amyloliquefaciens LL3 [24]. Wu et al. [23] also reported that by directing more carbon flux distribution toward glutamate synthesis by the presence of additives, γ-PGA production was increased. Those results suggested that elevating intracellular glutamate concentration may be an approach to improve γ-PGA production. Moreover, B. amyloliquefaciens LL3 is a glutamic acid-independent γ-PGA-producing strain, which suggests that intracellular glutamate is the only substrate for γ-PGA synthesis in this bacterium [5]. Therefore, it is tempting to speculate that elevated intracellular glutamate concentration facilitates γ-PGA production of LL3 more effectively than in glutamic acid-dependent γ-PGA-producing strains.
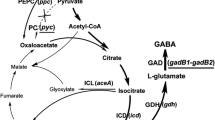
Besides rocR, there are several other genes known to participate in glutamate metabolism in Bacillus, which are summarized in Fig. 1 and Table 1. The gene rocG encodes the glutamate dehydrogenase (GDH) in B. subtilis, which is devoted to glutamate degradation rather than its synthesis [7]. The expression of rocG is controlled by the transcriptional activator RocR. RocG has another role as a regulator that inhibits GltC, which activates the gltAB operon encoding the glutamate synthase [10]. In B. subtilis, gudB is a second, cryptic glutamate dehydrogenase gene harboring an insertion of three amino acids with respect to the common ancestral GDH sequence [14]. In B. amyloliquefaciens LL3, however, the insertion does not exist (data not shown), suggesting its potential role in glutamate degradation together with RocG. The genes odhA, odhB and pdhD encode the 2-oxoglutarate dehydrogenase complex (ODHC) catalyzing the oxidative decarboxylation of 2-oxoglutarate to succinyl coenzyme A (succinyl-CoA) in B. subtilis [6]. By reducing the flux from 2-oxoglutarate to succinyl-CoA, the flux from 2-oxoglutarate to glutamate may be enhanced.

Postulated glutamate metabolism in B. amyloliquefaciens LL3 based on reports in B. subtilis 168. GudB and RocG are glutamate dehydrogenases. rocG is positively regulated by RocR. Alpha-oxoglutarate dehydrogenase complex (ODHC) mediates the conversion from 2-oxoglutarate to succinyl coenzyme A
The functions of the four genes (rocR, rocG, gudB and odhA) and their interactions led us to hypothesize that their deletions may elevate intracellular glutamate concentration, which facilitates γ-PGA production. With these expectations, we performed in-frame deletions of these four genes in B. amyloliquefaciens LL3 and examined the effects of their deletions on cell growth and γ-PGA production. By doing this we hope to identify more genetic targets for improvement of γ-PGA production.
Materials and methods
Strains and growth conditions
Strains used in this study are listed in Table 2. E.coli DH5α was used for plasmid construction and E. coli GM2163 was used to prepare the unmethylated plasmids. E. coli strains were grown in LB medium at 37 °C with aeration. The B. amyloliquefaciens LL3 strain is deposited in the China center for type culture collection (CCTCC) with accession number CCTCC M 208109 [9]. B. amyloliquefaciens strains were grown at 37 °C (except during gene deletion procedure) in LB or fermentation medium, which contains sucrose 50 g/L, (NH4)2SO4 2 g/L, MgSO4 0.6 g/L, KH2PO4 6 g/L, K2HPO4 14 g/L, and 2 mL mineral elements including 1 mM FeSO4·4H2O, CaCl2·2H2O, MnSO4·4H2O and ZnCl2 (pH 7.2). When required, media were supplemented with ampicillin (Ap 100 μg/mL) and chloramphenicol (Cm 5 μg/mL). Stock solutions (75 mg/mL) of 5-fluorouracil (5-FU) were prepared in DMSO and added into the media at a final concentration of 1.3 mM.
Plasmid construction
Primers used in this study are listed in Table 3. Homology arms for gene deletion were obtained using overlapping PCR and are 500–800 bp in length. A mutant allele of our target gene was obtained by overlapping PCR and then ligated to pKSV7-based counterselection plasmid pKSU. Using odhA deletion as an example, primers OdhAUP-F/OdhAUP-R and OdhADN-F/OdhADN-R were used to amplify the up- and down-stream homology arms. The two fragments were spliced in a subsequent PCR using primer pair OdhAUP-F/OdhADN-R. The resulting homology arms carried an in-frame deletion of the odhA gene and were ligated to pKSU. DNA polymerases, digestion enzymes and T4 DNA ligase were purchased from Takara (Dalian, China). PCR, enzymatic digestion and ligation reactions were performed as recommended by the enzymes suppliers. DNA fragments were analyzed on 0.8 % agarose gels and purified using an Axygen gel DNA recovery kit (Axygen, CA, USA).
Bacterial transformation
Competent E. coli cells were purchased from TransGen Biotech (TransGen, Beijing, China) and were transformed according to the manufacturer’s instructions. B. amyloliquefaciens LL3 and its derivatives were transformed using the high-osmolarity electroporation method as described previously [24].
Construction of recombinant strains
A upp-based markerless gene replacement method was used to perform in-frame deletion of the target genes [24]. LL3 ΔU carrying an in-frame deletion of the gene coding for the uracil phosphoribosyltransferase was used as the starting strain. Using odhA deletion as an example, deletion plasmid pKSU-ΔOdhA was transformed in LLΔU through electroporation. After plasmid integration/excision, double-crossover recombinants were selected with the aid of 1.3 mM 5-fluorouracil (5-FU) using the primer pair OdhAOUT-F/OdhAOUT-R. Primer nomenclatures for other gene deletions are similar. Mutant strains were verified using PCR and DNA sequencing. One correct colony carrying an in-frame deletion of odhA was designated LL3 ΔUA. Similarly, mutants carrying deletions for rocR, rocG and gudB were designated LL3 ΔUR, LL3 ΔUG and LL3 ΔUB, respectively. The amino acid residues retained in the mutants are shown in Table 1.
Fermentation in shake flasks and 5-L fermenters
For shake flask experiments, B. amyloliquefaciens LLΔU and its derivatives were first grown overnight in test tubes containing LB liquid medium, and then inoculated into 100 mL fresh fermentation medium in 500-mL shake flasks at an optical density at 600 nm (OD600) of approximately 0.05–0.1. The shake flasks were then grown at 37 °C for 48 h with an agitation speed of 200 rpm. Experiments were independently repeated at least three times and means and standard deviations were calculated. For fermentation in a 5-L jar fermenter (Bailun, Shanghai, China), cultures from the shake flasks grown overnight were used as seed cultures and diluted 10-fold into 3 L fresh fermentation medium. Fermentation was continued at 37 °C for 48 h with an agitation speed of 400 rpm. The pH was kept at 7.0 via automatic addition of 1 M NaOH. After 24 h, 50 mL fermentation broth was withdrawn every 6 h and centrifuged at 8,000g (4 °C) for 20 min. The cell pellet was washed three times with dH2O then dried and weighed to determine dry cell weight (DCW). Fourfold volume of cold anhydrous ethanol was added to the supernatant followed by incubation at 4 °C overnight. The precipitate was centrifuged at 4,000×g (4 °C) and then dialyzed against distilled water and lyophilized to obtain γ-PGA. [24].
Quantitative RT-PCR analysis
Bacillus amyloliquefaciens strains were grown to mid-log phase (approximately 20 h) in fermentation medium in shake flasks as described above. Cells were collected at 4 °C and the RNA was isolated using TransZol™ Up (TransGen, Beijing, China) according to the manufacturer’s instructions. cDNA was reverse transcribed using a GoScript™ reverse transcription system (Promega, Wisconsin, USA). Real-time PCR analysis for the target genes was performed using the SYBR Premix Ex Taq™ II (Takara, Dalian, China). Transcription levels of the target gene were normalized against the levels of rspU [8].
Preparation of cell-free B. amyloliquefaciens extracts and measurement of GDH and OGDH activity
Cultures (100 mL) grown under γ-PGA production conditions in shake flasks for 36 h were collected and washed four times with potassium phosphate buffer (50 mM, pH 7.5), and resuspended in 5 ml of the same buffer and disrupted by sonication for 15 min (500 W) on ice. The cell debris was removed by centrifugation at 4 °C for 10 min at 8,000g. Measurement of GDH and OGDH activities was performed as described by Krog et al. [11] with slight modifications. Briefly, the GDH assay mixture generally contained 100 mM glycine-KOH (pH 7.5), 200 mM L-glutamate, 10 mM MgSO4, and 0.2 mM NAD+; the OGDH assay mixture generally contained 50 mM Tris–HCl (pH 7. 5), 0.25 mM NAD+, 50 mM 2-oxoglutarate, 1 mM coenzymeA (CoA), 1 mM thiamine pyrophosphate, 3 mM l-cysteine-HCl, and 1 mM MgCl2. The mixtures were prewarmed to 37 °C in quartz cuvettes. After the addition of 0.1 ml cell extract, yielding a total volume of 1 ml, the production of NADH was monitored at 340 nm for 4 min. One unit of enzyme activity was defined as the amount of enzyme needed to produce 1 μmol NADH per minute under the conditions described above. The background activity from the cell extract was measured by adding all components but glutamate or 2-oxoglutarate, and the background activity was subtracted from the total activity. The total protein concentration was determined with the Bio-Rad protein assay (Bio-Rad).
Results
Construction of recombinant strains carrying deletions for rocR, rocG, gudB or odhA
The rocR mutant was previously described, and the other three mutants were constructed as described in the materials and methods section. For rocG and gudB deletion, approximately 50 % of the colonies subjected to PCR verification were shown to carry an in-frame deletion of the target genes (data not shown). For odhA deletion, however, only one mutant strain was identified among the 60 colonies that were subjected to PCR verification. The rocG gene was deleted in LL3 ΔUB to obtain the double-deletion carrying strain LL3 ΔUBG. LL3 ΔUGB was similarly constructed by deleting the gudB gene from LL3 ΔUG. The obtained mutants were streaked on LB agar and verified by PCR and DNA sequencing (Fig. 2a and data not shown).

Confirmation of the deletion-carrying mutants by PCR and phenotypes. a Chromosome structures of deletion-carrying strains were analyzed by agarose gel electrophoresis of PCR products generated with primers RocROUT-F/RocROUT-R (lanes 1–3); with primers RocGOUT-F/RocGOUT-R (lanes 4–6); with primers OdhAOUT-F/OdhAOUT-R (7–9); with primers GudBOUT-F/GudBOUT-R (lanes 10–12). M DNA marker, and the size of each band is reported on the left. Primer nomenclature is as in Table 2. The genotypes of strains are indicated on the top of each lane. Mutant strains carrying correct deletions would yield PCR products shorter than that of the wild type. Complementation strains would yield PCR products identical to that of the wild type. b Relative glutamate dehydrogenase (GDH) activities of the mutant strains. GDH activities of the rocR (LL ΔUR), rocG (LL ΔUG) and gudB (LL ΔUB) mutants were determined and normalized against that of the wild type (LL ΔU), which was set as 1. Values are an average from technical triplicates. c Relative 2-oxoglutarate dehydrogenase (OGDH) activity of the odhA (LL ΔUA) mutant. Values are an average from technical triplicates
Cell growth and γ-PGA production of the mutant strains in shake flasks
Growth profiles of the rocG, gudB, odhA and rocG/gudB (LL ΔUG, LL ΔUB, LL ΔUA and LL ΔUBG, respectively) mutants and the wild-type LL3ΔU were tested in LB medium as shown in Fig. 3a. The odhA mutant showed a severe growth defect, while others except the rocR mutant presented a growth pattern similar to the wild-type strain. The final dry cell weight (DCW) of the above mutants was also determined in fermentation medium at 48 h (Fig. 3b). Similar to that in LB medium, the odhA mutant showed the lowest DCW, while others showed a slight reduction compared with wild-type strain. The highest total γ-PGA production was found in LL3 ΔUBG which carries a double deletion for gudB and rocG, yielding 5.03 g/L γ-PGA compared with 3.64 g/L from the wild type (a 38.2 % increase). LL3 ΔUGB showed similar characteristics of growth and γ-PGA production with LL3 ΔUBG (data not shown), excluding the possibility of introduction of other mutations during strain construction. LL ΔUB showed a 15 % increase (4.245 g/L) in γ-PGA production, while LL ΔUG almost remained unchanged. Regardless of its deteriorated growth, the odhA mutant LL ΔUA produced 4.21 g/L γ-PGA, about 15 % more than the wild type. The specific production (the relative amount of γ-PGA per dry cell weight) for γ-PGA of the odhA mutant was 3.07 g/g DCW, presenting a 100 % increase compared with the wild type (1.5 g/g DCW), followed by the rocR mutant (2.79 g/g DCW, an 86 % increase). The data for the rocR mutant in shake flasks were reported previously and included here for comparison (Fig. 3a, b).

Characterization of the mutant strains in shake flasks. a Growth profiles of the rocG, gudB, odhA mutants and the rocG/gudB double mutant (LL ΔUG, LL ΔUB, LL ΔUA and LL ΔUBG, respectivley) in LB medium. Values were an average from biological duplicate set and standard deviations were essentially within 10 %, b cell growth and γ-PGA production of the mutants in fermentation medium at 48 h. c Growth and γ-PGA production of complementation strains for the rocR, odhA and gudB mutants (LL ΔUR, LL ΔUA and LL ΔUB, respectively) at 48 h. d Transcriptional levels of gene pgsB of the mutants (at 20 h). Values were an average from a biological triplicate set
Considering the noticeable changes in cell growth and γ-PGA production in the rocR, odhA and gudB mutants, their complementation strains were constructed. Our previous studies showed that overexpression of rocR in B. amyloliquefaciens LL3 caused severe growth defect [25]. Therefore, all the complementation experiments in this study were carried out by inserting the gene with its own promoter and terminator in its original position in the genome. Using rocR as an example, a fragment obtained by PCR with primer pair RocRUP-F/RocRDN-R using genomic DNA from LL ΔU as template was ligated to pKSU. The resulting complementation plasmid pKSU-RocR was used to transform LL3 ΔUR through electroporation. The rocR gene in pKSU-RocR was inserted in the genome of the rocR mutant strain by homologous recombination. Selection procedure for recombinant strains was similar to that of gene deletion as described previously. The resulting complementation strain for rocR was designated LL3 ΔUR-C. Complementation strains for rocG, gudB and odhA were similarly constructed. As shown in Fig. 3c, the phenotypes of the complementation strains resembled the wild-type strain in cell growth and γ-PGA production.
GDH and ODHC activities of the mutants
As a demonstration of roles of the target genes in glutamate metabolism, the GDH activities of the rocR, rocG and gudB mutants were determined and compared with the wild type. As shown in Fig. 2b, the rocR mutant (LL ΔUR) showed a 32 % decrease of GDH activity, and the rocG (LL ΔUG) and gudB (LL ΔUB) mutants showed ~50 % reduction. In the odhA (LL ΔUA) mutant, ODHC activity was very low, confirming the removal of the odhA gene (Fig. 2c).
Transcriptional levels of pgsB in the mutant strains
RocR and RocG are reported as transcriptional regulators. Therefore, we investigated whether transcription of pgsB (the first gene in the pgsBCA operon) was affected by the deletion of the rocR and rocG genes using qRT-PCR. As shown in Fig. 3d, no significant changes were observed in the mutant strains compared with the wild type at 20 h, which verified that the improved polymer production is not due to increased pgs operon transcription.
Further improvement of γ-PGA production using pH control strategy
Our previous report showed that the pH of the medium for the rocR mutant was lower than that of the wild type when fermented in shake flasks [24]; therefore, we performed fermentation of some of the mutants in fermenters with pH control. As shown in Fig. 4a, DCW of the odhA mutant (LL ΔUA) was obviously different from the other three mutants: it reached 2.81 g/L at 24 h, and dropped quickly thereafter, decreasing to 1.68 g/L at 48 h. The DCW of the wild-type strain showed a slight reduction after 24 h and was 3.09 g/L at 48 h. Compared with its growth in shake flasks, the odhA mutant showed obvious growth improvements in the fermenter, especially for the first 24 h. The final DCW at 48 h of the two culture conditions, however, showed no significant differences (1.37 g/L for shake flasks and 1.68 g/L for fermenters). Similar to results from shake flask experiments, the wild-type LL ΔU showed a slight advantage over the rocR (LL ΔUR) and gudB/rocG (LL ΔUBG) mutants in fermenters. DCW of LL ΔUR (2.56 g/L) and LL ΔUBG (2.77 g/L) in fermenters was improved compared with those in shake flask experiments (1.61 and 1.98 g/L, respectively).

Characterization of the mutant strains in fermenters. a DCW of the wild type, the rocR, odhA and gudB/rocG double mutants (LL ΔU, LL ΔUR, LL ΔUA and LL ΔUBG, respectivley) at different time points. b Absolute γ-PGA production of the mutants at different time points. All the values were means from a duplicate set, and standard deviations were essentially within 8 %
The highest gamma-PGA production for odhA mutant (LL ΔUA) was obtained at 42 h, reaching 4.6 g/L, a slight improvement compared with that in shake flask experiments (4.21 g/L at 48 h), while the wild-type strain produced 4.08 g/L γ-PGA at 48 h in fermenters. The gudB/rocG (LL ΔUBG) mutant produced 5.68 g/L γ-PGA, a slight increase compared with that for shake flask experiments (5.06 g/L). The γ-PGA production of the rocR mutant (LL ΔUR) was increased by 28 % in the fermenter compared with that in shake flask experiments (4.55 g/L), reaching 5.83 g/L, the highest among all the mutants tested (Fig. 4b).
Discussion
Gamma-PGA producers are usually divided into two groups according to their nutritional requirements: the glutamic acid-dependent bacteria and glutamic acid-independent bacteria. The former kind requires addition of L-glutamate in the medium to stimulate γ-PGA, and the latter does not. Wu et al. [23] reported that the addition of glycerol, Tween-80 and dimethyl sulfoxide (DMSO) increased the production of γ-PGA likely caused by depression of 2-oxoglutarate dehydrogenase and the enhancement of isocitrate dehydrogenase, which increased the flux from 2-oxoglutarate and isocitrate to glutamate, respectively. Enlightened by that, we attempted to enhance intracellular glutamate synthesis by genetically modifying related genes. Deletion of RocR (a regulator participating in glutamate degradation by activating the gene encoding glutamate dehydrogenase, rocG) was found to increase the flux from 2-oxoglutarate to glutamate, and then lead to increased synthesis of the other amino acids via transamination in B. subtilis 168 [13]. Besides rocR, rocG, gudB and odhA were also targeted in this study. We attempted to identify genes that might have a positive effect on γ-PGA production.
Unlike reports from B. subtilis 168 [14], the deletion of rocG (the gene encoding one of the glutamate dehydrogenase) in B. amyloliquefaciens LL3 seemed to confer no remarkable phenotype changes except the reduction of intracellular glutamate dehydrogenase (GDH) activity (Fig. 2b). In B. subtilis, RocG is the major glutamate dehydrogenase. The cryptic GudB (the second glutamate dehydrogenase) is only activated in the absence of RocG [10, 16]. In B. amyloliquefaciens LL3, however, the gudB gene lacks the insertion mutation found in B. subtilis. Therefore, GudB may play an equally or more important role as that of RocG. The gudB mutant (LL ΔUB) presented a slightly higher γ-PGA production and lower DCW (Fig. 3b), although not significant. What is worth noting is that although the rocR mutant (LL ΔUR) showed a higher GDH activity than LL ΔUB and LL ΔUG, it produced more γ-PGA (both total and specific production) than those two mutants (Figs. 2b, 3b). Transcription of the pgsBCA operon was not affected by the absence of RocR, as shown by qRT-PCR analysis (Fig. 3d). There seemed to be no clear relationships between GDH activities and γ-PGA production. The gudB/rocG double mutant (LL ΔUBG) presented the highest γ-PGA production in shake flask experiments among all the mutant strains constructed, and showed no obvious growth defect both in LB and fermentation medium, thus qualifying as a promising producer for further investigations.
In Corynebacterium glutamicum, the deletion of odhA (encoding α-oxoglutarate dehydrogenase complex that mediates the conversion from 2-oxoglutarate to succinyl coenzyme A) greatly induced the production of L-glutamate [1]. B. amyloliquefaciens LL3 deleted for odhA, however, showed severe growth defect in LB medium. The ODH complex is part of the citric acid (TCA) cycle which provides reducing power and anabolic biomass precursors to the cell. The complex may be involved in substrate channeling through the TCA cycle. Although not essential both in LB and fermentation medium, the absence of the odhA gene did affect cell growth. This may partly account for the low deletion rate (1/60) during its deletion. The growth of the odhA mutant was partly restored in the fermentation medium and its γ-PGA production was even a little higher than the wild type (Fig. 4a, b). The latter phenomenon may be explained by the fact that ODH complex is not directly involved in the biosynthesis of intracellular glutamate, which is the direct unit for γ-PGA production.
Our previous report showed that in shake flask experiments without pH control, the pH of the medium for the rocR mutant was lower than that of the wild type and we suggested that the drop of pH may be more detrimental for the mutant [24]. The restoration of growth defect of LL ΔUA in fermentation medium with phosphate buffer also indicated that pH control may facilitate the growth of the mutants. Enzyme production of the rocG mutant of B. subtilis was enhanced by the NH3-pH auxostat approach [14]. We therefore perform fermentation of LL ΔUR, LL ΔUBG and LL ΔUA in a 5-L fermenter using NaOH to keep pH stable at 7.0–7.2, expecting to enhance their growth and further improve their γ-PGA production. Unfortunately, LL ΔUA showed no improvements in DCW or γ-PGA yield (Fig. 4a). Lower DCW and quicker cell decline may contribute to the low γ-PGA yield. The LL ΔUBG mutant showed a 1.39-fold increase in γ-PGA production compared with the wild type, almost the same from shake flask experiments. A remarkable improvement was observed in LL ΔUR (Fig. 4b). Further optimization of fermentation parameters is needed to enhance the performance of the mutants, such as concentration of the nitrogen sources or pH maintenance using liquid ammonia.
Given the complicated nitrogen metabolism regulations in bacteria, modification of glutamate to improve cell growth and γ-PGA production needs tremendous work, and the number of genes selected for this study is in fact very limited. Enhancement of glutamate synthesis could also be achieved through introduction of certain genetic elements or replacement of regulatory elements (promoters for example) of genes responsible for glutamate synthesis, such as GltAB, and other regulators (GlnR for example) involved in nitrogen metabolism may also be targeted [7, 10], which will be our work in the future.
Conclusions
In summary, the present study attempted to uncover the effects of deletions of several genes involved in glutamate metabolism on γ-PGA production. Compared with the addition of additives in the fermentation medium, such as DMSO and glycerol, the engineered strains add no complexity or cost to fermentation process. The rocR mutants and the gudB/rocG double mutants showed great potential for industrial use. Our work identifies genes that can be targeted to improve γ-PGA production in B. amyloliquefaciens LL3, a strategy that may be also applicable in other γ-PGA producers to establish microbial cell factories with higher capacity.
References
Asakura Y, Kimura E, Usuda Y, Kawahara Y, Matsui K, Osumi T, Nakamatsu T (2007) Altered metabolic flux due to deletion of odhA causes L-glutamate overproduction in Corynebacterium glutamicum. Appl Environ Microbiol 73:1308–1319
Bajaj I, Singhal R (2011) Poly (glutamic acid)–an emerging biopolymer of commercial interest. Bioresour Technol 102:5551–5561
Bhat AR, Irorere VU, Bartlett T, Hill D, Kedia G, Morris MR, Charalampopoulos D, Radecka I (2013) Bacillus subtilis natto: a non-toxic source of poly-γ-glutamic acid that could be used as a cryoprotectant for probiotic bacteria. AMB Express 3:36
Candela T, Fouet A (2006) Poly-gamma-glutamate in bacteria. Mol Microbiol 60:1091–1098
Cao M, Geng W, Zhang W, Sun J, Wang S, Feng J, Zheng P, Jiang A, Song C (2013) Engineering of recombinant Escherichia coli cells co-expressing poly-γ-glutamic acid (γ-PGA) synthetase and glutamate racemase for differential yielding of γ-PGA. Microb Biotechnol 6:675–684
Carlsson P, Hederstedt L (1989) Genetic characterization of Bacillus subtilis odhA and odhB, encoding 2-oxoglutarate dehydrogenase and dihydrolipoamide transsuccinylase, respectively. J Bacteriol 171:3667–3672
Commichau FM, Herzberg C, Tripal P, Valerius O, Stülke J (2007) A regulatory protein-protein interaction governs glutamate biosynthesis in Bacillus subtilis: the glutamate dehydrogenase RocG moonlights in controlling the transcription factor GltC. Mol Microbiol 65:642–654
Feng J, Gao W, Gu Y, Zhang W, Cao M, Song C, Zhang P, Sun M, Yang C, Wang S (2014) Functions of poly-gamma-glutamic acid (γ-PGA) degradation genes in γ-PGA synthesis and cell morphology maintenance. Appl Microbiol Biotechnol 98:6397–6407
Geng W, Cao M, Song C, Xie H, Liu L, Yang C, Feng J, Zhang W, Jin Y, Du Y, Wang S (2011) Complete genome sequence of Bacillus amyloliquefaciens LL3, which exhibits glutamic acid-independent production of poly-γ-glutamic acid. J Bacteriol 193:3393–3394
Gunka K, Commichau FM (2012) Control of glutamate homeostasis in Bacillus subtilis: a complex interplay between ammonium assimilation, glutamate biosynthesis and degradation. Mol Microbiol 85:213–224
Krog A, Heggeset TM, Ellingsen TE, Brautaset T (2013) Functional characterization of key enzymes involved in L-glutamate synthesis and degradation in the thermotolerant and methylotrophic bacterium Bacillus methanolicus. Appl Environ Microbiol 79:5321–5328
Liu J, Ma X, Wang Y, Liu F, Qiao J, Li XZ, Gao X, Zhou T (2011) Depressed biofilm production in Bacillus amyloliquefaciens C06 Causes γ-poly-glutamic acid (γ-PGA) overproduction. Curr Microbiol 62:235–241
Manabe K, Kageyama Y, Morimoto T, Ozawa T, Sawada K, Endo K, Tohata M, Ara K, Ozaki K, Ogasawara N (2011) Combined effect of improved cell yield and increased specific productivity enhances recombinant enzyme production in genome-reduced Bacillus subtilis strain MGB874. Appl Environ Microbiol 77:8370–8381
Manabe K, Kageyama Y, Morimoto T, Shimizu E, Takahashi H, Kanaya S, Ara K, Ozaki K, Ogasawara N (2013) Improved production of secreted heterologous enzyme in Bacillus subtilis strain MGB874 via modification of glutamate metabolism and growth conditions. Microb Cell Fact 12:18
Mitsui N, Murasawa H, Sekiguchi J (2011) Disruption of the cell wall lytic enzyme CwlO affects the amount and molecular size of poly-γ-glutamic acid produced by Bacillus subtilis (natto). J Gen Appl Microbiol 57:35–43
Morimoto T, Kadoya R, Endo K, Tohata M, Sawada K, Liu S, Ozawa T, Kodama T, Kakeshita H, Kageyama Y, Manabe K, Kanaya S, Ara K, Ozaki K, Ogasawara N (2008) Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Res 15:73–81
Rajan YC, Inbaraj BS, Chen BH (2014) In vitro adsorption of aluminum by an edible biopolymer poly (γ-glutamic acid). J Agric Food Chem 62:4803–4811
Scoffone V, Dondi D, Biino G, Borghese G, Pasini D, Galizzi A, Calvio C (2013) Knockout of pgdS and ggt genes improves γ-PGA yield in B. subtilis. Biotechnol Bioeng 110:2006–2012
Shih IL, Van YT (2001) The production of poly (γ-glutamic acid) from microorganisms and its various applications. Bioresour Technol 79:207–225
Smith K, Youngman P (1992) Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spoIIM gene. Biochimie 74:705–711
Su Y, Li X, Liu Q, Hou Z, Zhu X, Guo X, Ling P (2010) Improved poly-gamma-glutamic acid production by chromosomal integration of the Vitreoscilla hemoglobin gene (vgb) in Bacillus subtilis. Bioresour Technol 101:4733–4736
Sung MH, Park C, Kim CJ, Poo H, Soda K, Ashiuchi M (2005) Natural and edible biopolymer poly-γ-glutamic acid: synthesis, production, and applications. Chem Rec 5:352–366
Wu Q, Xu H, Shi N, Yao J, Li S, Ouyang P (2008) Improvement of poly (gamma-glutamic acid) biosynthesis and redistribution of metabolic flux with the presence of different additives in Bacillus subtilis CGMCC 0833. Appl Microbiol Biotechnol 79:527–535
Zhang W, Gao W, Feng J, Zhang C, He Y, Cao M, Li Q, Sun Y, Yang C, Song C, Wang S (2014) A markerless gene replacement method for B. amyloliquefaciens LL3 and its use in genome reduction and improvement of poly-γ-glutamic acid production. Appl Microbiol Biotechnol 98:8963–8973
Zhang W, Xie H, He Y, Feng J, Gao W, Gu Y, Wang S, Song C (2013) Chromosome integration of the Vitreoscilla hemoglobin gene (vgb) mediated by temperature-sensitive plasmid enhances γ-PGA production in Bacillus amyloliquefaciens. FEMS Microbiol Lett 343:127–134
Acknowledgments
This work was supported by National key Basic Research Program of China (“973”-Program) 2012CB725204, National High Technology Research and Development Program of China (“863”-Program) 2012AA021505, Natural Science Foundation of China Grant Nos. 31170030, 31300032 and 51073081, Project of Tianjin, China (13JCZDJC27800, 13JCYBJC24900, 13JCQNJC09700 and 14ZCZDSF00009).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhang, W., He, Y., Gao, W. et al. Deletion of genes involved in glutamate metabolism to improve poly-gamma-glutamic acid production in B. amyloliquefaciens LL3. J Ind Microbiol Biotechnol 42, 297–305 (2015). https://doi.org/10.1007/s10295-014-1563-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1563-8