
Abstract
Based on the original thermostable alpha-amylase gene from Bacillus licheniformis, two amino acids were site-directed mutagenised by polymerase chain reaction to obtain a new gene. This gene, with Leu134→Arg and Ser320→Ala, was substituted for acid-resistant capability previously. To favor purification of the product, high-level expression and secretion of mature, authentic and stable recombinant mutagenised alpha-amylase were achieved with protease-deficient strain Bacillus subtilis WB600 as the host. The recombinant mutagenised alpha-amylase with the activity of 4,700 U/mL was then purified by ammonium sulphate fractionation, anion exchange and gel filtration, consecutively. By multi-step purification, the specific activity of the recombinant protein was up to 916.7 U/mg with a 187.1-fold purification. The mutagenised protein was found to be more acid resistant than the native protein. The optimum pH and stable range of pH with the mutagenised protein was 4.5 and 4.0 to 6.5, respectively, compared with pH 6.5 and 5.5 to 7.0 as the favorite pH and pH stability range of the native protein.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alpha-amylase (α-1,4-glucan 4-glucanohydrolase, EC 3.2.1.1) catalyses the hydrolysis of the α-1,4 glycosidic linkages of starch, found in amylose and amylopectin. It is currently used in such a broad array of industrial applications as starch hydrolysis for the production of ethanol and high fructose corn syrup, starch soil removal in laundry washing powders and dish-washing detergents, textile de-sizing, the production of modified starches, baking, hydrolysis of oil-field drilling fluids and paper recycling (Richardson et al. 2002). Alpha-amylase isolated from the ubiquitous mesophilic soil bacterium Bacillus licheniformis was widely used in these areas (Saito 1973; Yuuki et al. 1985; Matsuzaki et al. 1974). This enzyme operates optimally at 90°C and pH 6, and it requires addition of calcium ions for its thermostability (Violet and Meunier 1989). Although B. licheniformis alpha-amylase (BLA) was utilised widely for its thermostable character (Machius et al. 2003), it is sensitive to acidic circumstances, and this could result in the loss of its hydrolysis ability (Lee et al. 2006). Because several industrial processes take place at lower pH values than those where the alpha-amylase perform optimally (Nielsen and Borchert 2000), this entails significant raw material and processes operating costs for pH adjustments at a large scale (Crabb and Shetty 1999).
In the case of alpha-amylase, most of the protein engineering work has been devoted to its operative pH range (Nielsen et al. 1999; Shaw and Bott 1996), based on the thermostable property considerations. Therefore, to improve the acid-resistant property of BLA, in our previous research, the BLA gene that codes for Leu134 and Ser320 was modified to code for Arg and Ala, respectively (Cai et al. 2005). Then, the mutagenised BLA was expressed in Bacillus subtilis DB403 under the control of the promoter with sacB gene. However, recombinant protein has low yield and low activity while its enzyme properties were not analysed, so that whether the mutagenised gene could resist acid was not sure.
Although the B. subtilis expression–secretion system was an effective expression system, there was still a major limitation in that B. subtilis produced and secreted high levels of extracellular proteases, which degraded the secreted foreign proteins (Wong et al. 1986). In a previous experiment, the B. subtilis DB403 deficient in three major extracellular proteases was used as the host, but the expressed mutagenised BLA was limited by the remaining proteases of B. subtilis DB403. To overcome the degradation problem caused by B. subtilis proteases, B. subtilis WB600, which was deficient in six extracellular proteases, was used for the expression of the heterologous genes (Wu et al. 1991). In addition, the vector pWB980 with a high plasmid copy number (121 copies per cell) contained a P43 promoter, a sacB signal sequence, a multiple cloning site and a kanamycin resistance marker from B. subtilis (Wu and Wong 1999). The P43 promoter was a strong and constitutively expressing promoter for direction with the transcription of the amylase genes in B. subtilis during growth. That the recombinant protein secreted into the culture broth was allowed by the sacB signal sequence.
Based on these reasons, the mutagenised gene was inserted into the B. subtilis expression plasmid pWB980 for high-yield recombinant protein expressed in B.subtilis WB600 in the present study. High productivity with little extracellular protein was advantageous to the downstream purification of the target protein. Meanwhile, high productivity of the target protein was convenient for the determination of enzyme activity. The properties of the purified mutagenised BLA was analysed exactly, when native BLA characteristics was used as a control. Detailed characterisation was exhibited to determine whether some superior properties such as acid resistance were expressed in the mutagenised protein.
Materials and methods
Recombinant plasmid construction
pUAM was constructed by inserting a 1.9-kb deoxyribonucleic acid (DNA) fragment that contained a BLA gene with signal peptide to pUC19, while pUAMD was constructed by inserting a 1.9-kb DNA fragment that contained a mutagenised BLA gene with a signal peptide to pUC19. DNA fragments encoding for mature peptide of native BLA (amy) and mutagenised BLA (amyd) were amplified using the plasmid pUAM and pUAMD as templates with a pair of polymerase chain reaction (PCR) primers, respectively, primer 1: 5′-CCCAAGCTTGCAAATCTTAATGGGACGCT-3′ and primer 2: 5′-CGGGGTACCAGAAACTTGTATTTAACTTT-3′. amy and amyd had a HindIII site at the 5′ end and a BamHI site at the 3′ end. The DNA fragments digested by HindIII and BamHI were then cloned into HindIII–BamHI-linearised pWB980, to give pWB-amy and pWB-amyd using standard procedures (Sambrook et al. 1989). The recombinant plasmids were transformed into the expression host, B. subtilis WB600.
Transformation of B. subtilis
Competence media (GMI and GMII) and minimal media were prepared and used as described (Yasbin et al. 1975). Cells were grown in GMI overnight at 30°C. To 45 mL GMI medium, 5 mL overnight culture grown in GMI was added and incubated at 37°C with shaking at 250 rpm for 4 h. Five millilitres of the early stationary phase culture was then mixed with 45 mL GMII medium, and the incubation continued at 37°C for 1 h. DNA samples (about 1–3 μg) and 0.5 mL competent cells were mixed and incubated at 37°C for 30 min. The cells were plated on a Luria–Betani (LB)/agar plate containing kanamycin (20 μg/mL) and incubated at 37°C for 12 h. The vector without inserts were transformed into the strains of B. subtilis WB600 and used as control.
Expression of recombinant protein AMY and AMYD
B. subtilis WB600 cells harbouring pWB-amy and pWB-amyd were cultivated overnight and 50 times diluted in LB broth (50 mL) supplemented with kanamycin (20 μg/mL), and the cells were grown for 36 h at 37°C. The liquid culture was centrifuged at 10,000 rpm for 10 min to remove cells. Culture supernatant was collected for activity determination and sodium dodecyl sulphate (SDS)–polyacrylamide gel electrophoresis (PAGE) analysis of recombinant protein.
Sodium dodecyl sulphate–polyacrylamide gel electrophoresis
The sample for SDS-PAGE was mixed with 2× SDS loading buffer (100 mmol/L Tris–HCl, pH 6.8, 200 mmol/L dithiothreitol, 0.4 g/L SDS, 0.02 g/L bromophenol blue and 20% [v/v] glycerol) in 1:1 (v/v) ratio. Then, the mixture was boiled for 5 min and centrifuged for 10 min at 12,000 rpm. The supernatant was used for SDS-PAGE (5% [v/v] stacking gel and 10% [v/v] resolving gel). Proteins were visualised with Coomassie Brilliant Blue.
Purification of recombinant protein AMY and AMYD
Culture supernatant (100 mL) was salted out with ammonium sulphate at 70% saturation. The precipitate was centrifuged at 6,000 rpm for 10 min and dissolved into 100 mL of 20 mmol/L Tris–HCl buffer (pH 7.0) and then applied onto a diethylaminoethyl-Sepharose fast flow column (2.5 × 20 cm) equilibrated with 20 mmol/L Tris–HCl buffer (pH 7.0) and eluted with 20 mmol/L Tris–HCl buffer (pH 7.0) containing a linear gradient from 0 to 1 mol/L NaCl solution. The active fractions were loaded on a Sephadex G-75 gel column (1.6 × 80 cm) equilibrated with 20 mmol/L phosphate buffer (pH 7.0) in 0.15 mol/L NaCl solution, then eluted by the same buffer (pH 7.0) at a rate of 0.5 mL/min. The active fraction obtained from the gel filtration column was dialysed against 20 mmol/L phosphate buffer (pH 7.0). One-millilitre fractions were collected to determine enzyme activity. Protein was monitored as described by Lowry et al. (1951).
Analysis of the enzyme activity
The definition of alpha-amylase is described below. One unit of enzyme is the amount of amylase needed to complete the liquefaction of starch into dextrin per minute at 70°C and pH 6.0. The measurement was done according to Chinese Industrial Standard (QB/T 2306-97). The calculation of the enzyme activity was based on the formula below. X = c × n × 16.67, where X is the enzyme activity of the sample (U/mL), c is the concentration of the control enzyme (U/mL) corresponded with the absorbance and n is the dilution fold.
Enzyme activity assay
The optimal temperature for amylase activity was determined by assaying activity between 30 and 100°C. Thermostability of the amylase was performed by maintaining the purified enzyme solution in water bath at different temperature (40, 60, 80 and 100°C) for 20, 40, 60, 80, 100 and 120 min, respectively, and then running the activity assay. Measurement of optimum pH for amylase activity was carried out by running the activity assay between the pH range of 3.0 and 7.0. Under several pH conditions (3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5 and 7.0), the pH stability was determined by incubating purified enzyme in a water bath at 70°C for 1 h, and then the residual enzyme activity was showed to analyse the pH stability All measurements were undergone at least in triplicate. In addition, to identify the influence of metal ions on the enzyme activity, the amylase was dialysed against 10 mmol/L Tris–HCl buffer (pH 7.0) containing 1 mmol/L ethylenediamine tetraacetic acid to remove excess ions first, and then the amylase activity was measured at pH 6.0 and 70°C for 1 h with 1 mmol/L various metal ions (Cu2+, Zn2+, Ba2+, Mg2+, Mn2+, Ca2+, Co2+, Cs2+, Cd2+, Fe2+, Hg2+, Ni2+ and Sr2+). Then thermostability of the enzyme in the presence and absence of various metal ions was investigated by incubating it with different concentration of the metal ion solution (1–6 mmol/L and a control group without metal ions present) at 95°C for 1 h.
The determination of dextrose equivalents value
The dextrose equivalents (DE) measurement was done according to Chinese Industrial Standard (QB 1216-91). The calculation of DE value was based on the formula below, \(DE = \frac{{1 \times V{\text{1}} \times 10}}{{V{\text{2}} \times m \times G}}\), where DE is the DE value of the sample, V 1 is the volume of standard glucose solution during titration (1 g/L), V 2 is the volume of sample, m is the mass of sample and G is the content of solid property.
Application of recombinant protein AMY and AMYD
Purified protein was added as quantity of 30 U/(g starch) into 100 mL starch solution (20%, w/v), which was adjusted to pH 4.5. After incubation at 95°C for 90 min, pH was adjusted to 2.0 using 1 mol/L HCl to inactivate enzymes for termination reaction. The starch solution was quickly cooled to room temperature. The transmittance was measured using distilled water as control at the wavelength of 640 nm. Subsequently, the solution was added with adequate NaOH solution to adjust pH to 4.5, and then DE value was measured.
Computer modeling methods
To obtain the theoretical structure of mutant (L134R and S320A) and native BLA, we built models using a public website, Swiss-Model (Peitsch et al. 1995, 1996; Peitsch 1996; Guex and Peitsch 1997), using the coordinates of the wide type from BLA (1VJS pdb entry) as a template, which was received from the Protein Databank (PDB; Bernstein et al. 1977).
Results
Cloning, sequencing and characterisation of the amy gene and amyd gene from the insert
The amplified amy gene and amyd gene by PCR was cloned into HindIII–BamHI restriction sites of the pWB980 vector to construct recombinant plasmids pWB-amy and pWB-amyd. Competent B. subtilis WB600 cells were transformed with the recombinant plasmids and screened on a LB/agar plate containing kanamycin (20 μg/mL). Confirmed by nucleotide-sequencing analysis, the mutagenesis of two sites, Leu134→Arg and Ser320→Ala, were obtained. The DNA fragments containing the amy gene and amyd gene comprised 1,449 nucleotides, respectively, which encoded a protein of 483 amino acids whose molecular weight was calculated as 53,130 Da (Fig. 1).

Alignment of mature peptide between native protein AMY and mutagenised protein AMYD. Different amino acids (Leu134→Arg and Ser320→Ala) substituted are shown in grey
Expression of recombinant protein AMY and AMYD in B.subtilis
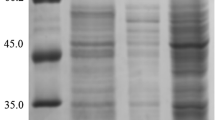
For the pWB-amy and pWB-amyd expression vectors, the activity of native protein AMY and mutagenised protein AMYD in the supernatant of the culture medium reached a maximum of 4,500 and 4,700 U/mL, respectively, after 36 h at 37°C. AMY and AMYD were analysed by SDS-PAGE (Fig. 2). In SDS-PAGE, the molecular weight of the expressed protein was about 53 kDa, which was in good agreement with that (53,130 Da) calculated from the amino acid sequence.

SDS-PAGE analysis of proteins secreted by B. subtilis WB600. Lane M, molecular weight markers; lane 1, culture supernatant of B. subtilis WB600 harbouring pWB-amy; lane 2, purified native protein AMY; lane 3, culture supernatant of B. subtilis WB600 harbouring pWB-amyd; lane 4, purified mutagenised protein AMYD; lane 5, culture supernatant of B. subtilis WB600 harbouring pWB980
Purification of recombinant protein
A general summary of the alpha-amylase purification protocol was presented in Table 1. It was evident that being subjected to ammonium sulphate fractionation, anion exchange and gel filtration, consecutively, the crude supernatant of AMY and AMYD yielded a highly enriched alpha-amylase preparation. AMY was purified 202.5-fold with a specific activity of 836.4 U/mg, while AMYD was purified 187.1-fold with a specific activity of 916.7 U/mg. The molecular weight of the purified enzymes, which appeared as a single band on SDS-PAGE, was found to be 53 kDa (Fig. 2).
AMY and AMYD activity assay
The purified AMYD displayed its optimum activity at 95°C. Besides it, remarkable thermal stability was also shown in AMYD. Rather, little loss of the activity was observed at 40, 60 and 80°C after incubating for 2 h. Around 65% of the enzyme activity was still detectable after incubating in a boiling water bath for 1 h. AMY has the same characters in optimal temperature and thermal stability. Thus, there was no change in the thermostable property after mutation.
The optimum pH for AMYD and AMY was 4.5 and 6.5, respectively (Fig. 3). When the pH was below 4.5, compared with AMY, which had a rapid decline with the activity, AMYD could maintain its activity strongly. Even AMYD had 50% of the highest activity at pH 3.5. It was obvious that AMYD also had a well activity in an acidic environment, where pH ranged from 4.0 to 6.5 (Fig. 4). On the contrary, AMY showed activity stability in a pH range between 5.5 and 7.0. Acid stability of AMYD was rather trustful even if pH was lower than 4.0, and only minor activity was lost. In contrast, AMY was sensitive to acidic circumstance and showed a decreasing activity when the pH was adjusted to below 5.0. It was concluded that AMYD was ready for use in liquefaction processes, which would occur in low pH value.

Detemination of recombinant protein pH optima. Relative enzyme activities (% of maximum) of AMY and AMYD at different pH levels are shown. Enzyme activity was assayed by incubating the enzyme at different pH levels for 5 min at 70°C. Filled squares, AMY; empty squares, AMYD

Determination of recombinant protein pH stability. The enzymes AMY and AMYD were incubated in buffers of differing pH for 60 min at 70°C before measuring the residual activity. The counterpart incubated for 5 min at 70°C was used as a control. Filled squares, AMY; empty squares, AMYD
For the investigation of the impact of different metal ions, Cu2+, Zn2+, Ba2+, Mg2+, Mn2+, Ca2+, Co2+, Cs2+, Cd2+, Fe2+, Hg2+, Ni2+ and Sr2+ at 1 mmol/L was incubated with purified AMYD and AMY. The addition of some metal ions (Table 2) caused enzyme inhibition. Hg2+ completely inhibited enzyme activity at 1 mmol/L. Cu2+, Zn2+, Ba2+, Mg2+, Mn2+, Cd2+ and Ni2+, respectively, showed 49, 37, 28, 5, 15, 48 and 24% inhibition to the AMYD activity, respectively. Similar effects on the AMY were obtained when the same metal ions were added separately. Meanwhile, Ca2+ displayed the highest thermostability-assistant property at a concentration of 2 mmol/L for AMYD and AMY. However, when the concentration exceeded 2 mmol/L, it showed a decrease with activity as a result. As the concentration of the other metal ions was increased, there was no significant influence on the activity of AMYD and AMY. The results indicated that mutation sites had no change to the effects of metal ions on the alpha-amylase activity.
Experiment for application
The purified AMY and AMYD were applied into starch hydrolysis assays. At pH 4.5 and 95°C, the DE value of the liquefaction solution caused by AMYD was higher than the counterpart control of AMY significantly (Fig. 5). The aggregation of the impurity in the liquefaction solution was reflected by the transmittance directly. The better aggregation, the higher the transmittance was. Then, transmittance was another parameter for the liquefaction. As shown in Fig. 6, the transmittance of liquefaction solution with AMYD was higher than the control one. It was concluded that the aggregation degree of AMYD was better than AMY at pH 4.5 and 95°C. All the results showed that compared with AMY, excellent acid-resistant ability was existed in AMYD.

The determination of the DE value. The DE of liquefied syrups was determined by measuring the quantity of reducing sugars (as glucose) by the neocuproine procedure. The amount of glucose in the sample was determined by comparison to a known glucose standard (1 g/L). Filled squares, AMY; empty squares, AMYD

The determination of transmittance for liquefaction solution. After reaction was finished, the transmittance was measured using distilled water as control at the wavelength of 640 nm. Filled squares, AMY; empty squares, AMYD
Discussion
A thermophilic and acid-resistant enzyme was obtained by expression with mutagenised gene in B. subtilis WB600. Compared with recombinant protein expressed in harbouring pHPSM plasmid of the recombinant B. subtilis DB403 (Cai et al. 2005), this recombinant protein had a 1.5-fold higher activity. The specific activity of this new enzyme was then up to 916.7 U/mg after purification with ammonium sulphate fractionation, anion exchange and gel filtration, consecutively. Incubated at 100°C for 60 min, 70% enzyme activity could still be maintained showing there were no obvious differences in thermal stability between the native enzyme and the mutagenised enzyme. However, the mutagenised enzyme was more acid resistant. Its optimum pH was 4.5, and the stable pH range was 4.0 and 6.5, which was superior to the native enzyme with the optimum pH of 6.5 and the stable pH range of 5.5 to 7.0. Meanwhile, at the starch hydrolysis experiment, mutagenised enzyme was more effective than the native enzyme under the circumstance of pH 4.5 and temperature 95°C.
As a host for heterologous expression, B. subtilis, which was an aerobic and mesophilic bacterium, has many attractive features (Wong 1995). These features included the ability to secrete extracellular proteins, ease of genetic manipulation and fast growth. However, one major problem was the presence of high levels of extracellular proteases. Although we used a mutant deficient in three major extracellular proteases (Cai et al. 2005), the expressed protein was truncated by the remaining proteases of B. subtilis DB403. To solve the degradation caused by B. subtilis proteases, we used B. subtilis WB600, which was deficient in six proteases for enhancing the effect without overproducing extracellular proteases. Moreover, it was in the interest of the biotechnological application and industry to seek new strong promoters. A lot of information of B. subtilis promoter had been acquired, and several of them had been exploited to be used as a control element in the construction of expression vector in B. subtilis, of which some constitutive promoters, such as the P43 promoter, was widely investigated and characterised (Hartl et al. 2001; Meijer and Margarita 2004; Zhang et al. 2005). Amongst them, the P43 promoter was used as a common control element in the construction of expression vector in B. subtilis and was considered as a strong promoter (Zhang et al. 2005). The B. subtilis WB600 and vector pWB980 with P43 promoter were chosen in the present research owing to their superior properties. The mutagenised gene was cloned into pWB980 and expressed in B. subtilis WB600 for high-yield recombinant protein. A high-level expression of mutagenised BLA was achieved in the recombinant B. subtilis with the activity 2.5 times to that of the previous one (Cai et al. 2005). The target protein was convenient to be purified and obtained because of fewer influence caused by that fewer species of the extracellular protein secreted by B. subtilis WB600.
Recently, although significantly different enzyme properties were researched by a collection of wild-type alpha-amylase from different environments, no single enzyme could possess all the desired properties for the targeted industrial application process where there was considerable need for thermophilic alpha-amylase expressed at high levels in the acid environment (Richardson et al. 2002). As a powerful tool to improve the properties of proteins, directed evolution was utilised widely (Kuchner and Arnold 1997). Protein engineering work was applied to the BLA to improve its thermal stability by rational protein engineering (Svensson and Sogaard 1992; Svensson 1994; Declerck et al. 1995; Declerck et al 1997; Declerck et al. 2000; Igarashi et al. 1998). Lately, the tolerance of the BLA toward low pH was enhanced by directed evolution (Shaw et al. 1999). The performance of profile of BLA has been improved by site-directed mutagenesis to change the dynamics of the active site residues and the electrostatics of the active site (Nielsen et al. 1999; Nielsen et al. 2001).
It was generally assumed that the stability of proteins was not affected by the amino acid substitutions on the surface with large amounts. This was because most surface residues are involved in only a few often transient interactions. Yet, we and others (Arnold and Volkov 1999; Giver et al. 1998; Ness et al. 1999; Schmidt-Dannert and Arnold 1999; Coco et al. 2001) have demonstrated that directed evolution is an efficient tool for the improvement of enzymes. The engineering work on BLA (Cai et al. 2005) showed that a few point mutations at the surface are effective to drastically increase the resistance against acidic conditions. In our previous paper, residues Leu134 and Ser320 were replaced by Arg and Ala in BLA, respectively. Surprisingly, the mutagenised enzyme showed outstanding properties of well endurance in acidic environment.
It was shown (Fig. 7) that the alpha-amylase consisted of three domains, which was the same to the result analysed by X-ray crystallographic studies (Nielsen et al. 1999). Domain A, which is a central (α/β)8 triosephosphateisomerase (TIM) barrel, forms the core of the molecule, consists of residues 4–103 and residues 203–396 and contains three active site residues Asp231, Glu261 and Asp328 (BLA numbering). Asp231 and Glu261 are believed to be the two catalytic groups. Asp231 is the catalytic nucleophile, while evidence has been presented for Glu261 being the catalytic hydrogen donor (Mc Carter and Withers 1996; Uitdehaag et al. 1999). The third essential acid (Asp328) is believed to assist catalysis by hydrogen bonding to the substrate and by elevating the pK a of Glu261 (Klein et al. 1992; Knegtel et al. 1995; Strokopytov et al. 1995; Uitdehaag et al. 1999). The active site is located in a cleft at the interface between domains A and B, where the C termini of the β-strands and loops joining the β-strands to the α-helix in the TIM barrel are found (Nagano et al. 2001). Domain B, formed by residues 104–202, is a protrusion between the third strand and the third helix of the TIM barrel (MacGregor 1993) and forms an irregular β-like structure. The size and structure of domain B varies substantially amongst the various members of the alpha-amylase family. This domain is probably responsible for the differences in substrate specificity and stability amongst the alpha-amylases (Svensson 1994). Domain C formed by residues 397–483 is located roughly at opposite sides of this TIM barrel to domain B. It forms a Greek key motif and contains the C terminus.

The position of the mutations further away from the active site in the structure of BLA. The structure was yielded by homology modelling based on the structure of BLA (1VJS pdb entry) using the Swiss-Model server (http://www.expasy.ch/swissmod)
Alpha-amylase catalysis is thought to be limited by the deprotonation of the nucleophile (D231) at low pH and the protonation of the hydrogen donor (E261) at high pH (Fang and Ford 1998; Qian et al. 1994; Strokopytov et al. 1995; Krengel and Dijkstra 1996). The protonation state of D231 is therefore at least partly responsible for the low activity at acidic pH. However, others (Nielsen et al. 1999) concluded that the mutations aimed at the change of the pH-dependent properties of enzymes by changing the hydrogen-bond interactions and the solvent accessibility of the active site residues should not be close to a catalytic residue. Mutations (L134R and S320A) outside the active site (Fig. 7) were designed away from the active site to influence the pK a values. Significant changes with the outstanding acid-resistant property were achieved in the mutagenised alpha-amylase by site-directed mutagenesis. We speculated that these mutations changed the net charge on the mutate residues, and more than two heavy charges were introduced or removed in the active site electrostatics, and then the pK a values of E261 and/or D328 was disturbed.
In addition, enzymes could be stabilised by stabilising the folding state, destabilising the unfolded state and altering the kinetics of unfolding in the many researches (Shaw and Bott 1996). And entropy increasing (ΔS) was an important driving force in protein folding. It was deduced that mutations that decreased the entropy of the system of the unfolded protein or increased the entropy of the system of the folded protein would lead to a larger ΔS for folding, which could stabilise the structure without complementary enthalpic contributions. It was of importance to protein engineering by unfolding and folding kinetics manipulation, and the insights to the structures of folding transition states are very valuable.
To test this hypothesis, we are currently using the x-ray diffraction technique to study the spatial structure of the mutant (L134R and S320A) and native BLA. With comparing the three-dimensional structures of them, we would accurately achieve the factors affecting the acid stability and catalytic activity on the structural level.
A thermophilic high-activity enzyme with excellent acid resistance obtained in this research is beneficial to starch hydrolysis in industrial production because of a convenient manipulation and a lower cost. The result is paved for substitution of the alpha-amylase used in current ferment and commercial acid-resistant alpha-amylase production in the future. Moreover, this study illuminates that protein engineering can be extended to modify other residues, and it also can improve their catalysis ability, with important implications for the starch industrial process.
References
Arnold FH, Volkov AA (1999) Directed evolution of biocatalysts. Curr Opin Chem Biol 3:54–59
Bernstein FC, Koetzle TF, Williams GJ, Meyer Jr EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M (1977) The Protein Databank. A computer-based archival file for macromolecular structures. Eur J Biochem 80:319–324
Cai H, Chen ZJ, Du LX, Lu FP (2005) Expression and secretion of an acid-stable α-amylase gene in Bacillus subtilis by sacB promoter and signal peptide. Biotechnol Lett 27:1731–1736
Coco WM, Levinson WE, Crist MJ, Hektor HJ, Darzins A, Pienkos PT, Squires CH, Monticello DJ (2001) DNA shuffling method for generating highly recombined genes and evolved enzymes. Nat Biotechnol 19:354–359
Crabb WD, Shetty JK (1999) Commodity scale production of sugars from starches. Curr Opin Microbiol 2:252–256
Declerck N, Joyet P, Trosset JY, Garnier J, Gaillardin C (1995) Hyperthermostable mutants of Bacillus licheniformis α-amylase: multiple amino acid replacements and molecular modelling. Protein Eng 8:1029–1037
Declerck N, Machius M, Chambert R, Wiegand G, Huber R, Gaillardin C (1997) Hyperthermostable mutants of Bacillus licheniformis alpha-amylase: thermodynamic studies and structural interpretation. Protein Eng 10:541–549
Declerck N, Machius M, Wiegand G, Huber R, Gaillardin C (2000) Probing structural determinants specifying high thermostability in Bacillus licheniformis alpha-amylase. J Mol Biol 301:1041–1057
Fang TY, Ford C (1998) Protein engineering of Aspergillus awamori glucoamylase to increase its pH optimum. Protein Eng 11:383–388
Giver L, Gershenson A, Freskgard PO, Arnold FH (1998) Directed evolution of a thermostable esterase. Proc Natl Acad Sci USA 95:12809–12813
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723
Hartl B, Wehrl W, Wiegert T, Homuth G, Schumann W (2001) Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol 183:2696–2699
Igarashi K, Hatada Y, Ikawa K, Araki H, Ozawa T, Kobayashi T, Ozaki K, Ito S (1998) Improved thermostability of an arginine-glycine residue is caused by enhanced calcium binding. Biochem Biophys Res Commun 248:372–377
Klein C, Hollender J, Bender H, Schulz GE (1992) Catalytic center of cyclodextrin glycosyltransferase derived from X-ray structure analysis combined with site-directed mutagenesis. Biochem 31:8740–8746
Knegtel RM, Strokopytov B, Penninga D, Faber OG, Rozeboom HJ, Kalk KH, Dijkhuizen L, Dijkstra BW (1995) Crystallographic studies of the interaction of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 with natural substrates and products. J Biol Chem 270:29256–29264
Krengel U, Dijkstra BW (1996) Three-dimensional structure of endo-1,4-beta-xylanase I from Aspergillus niger: molecular basis for its low pH optimum. J Mol Biol 263:70–78
Kuchner O, Arnold FH (1997) Directed evolution of enzyme catalysts. Trends Biotechnol 15:523–530
Lee S, Oneda H, Minoda M, Tanaka A, Inouye K (2006) Comparison of starch hydrolysis activity and thermal stability of two Bacillus licheniformis α-amylases and insights into engineering-amylase variants active under acidic conditions. J Biol Chem 139:997–1005
Lowry OH, Rosbrough NL, Farr AL (1951) Protein measurement with folin phenol reagent. J Biol Chem 193:265–275
MacGregor EA (1993) Relationships between structure and activity in the α-amylase family of starch-metabolising enzymes. Starch 7:232–237
Machius M, Declerck N, Huber R, Wiegand G (2003) Kinetic stabilization of Bacillus licheniformis α-amylase through introduction of hydrophobic residues at the surface. J Biol Chem 278:11546–11553
Matsuzaki H, Yamane K, Yamaguchi K, Nagata Y, Maruo B (1974) Hybrid alpha-amylases produced by transformants of Bacillus subtilis. I. Purification and characterization of extracellular alpha-amylases produced by the parental strains and transformants. Biochim Biophys Acta 365:235–247
McCarter JD, Withers SG (1996) Unequivocal identification of Asp-214 as the catalytic nucleophile of Saccharomyces cerevisiae alphaglucosidase using 5-fluoroglycosyl fluorides. J Biol Chem 271:6889–6894
Meijer WJJ, Margarita S (2004) Relevance of UP elements for three strong Bacillus subtilis phage phi 29 promoters. Nucleic Acids Res 32:1166–1176
Nagano N, Porter CT, Thornton JM (2001) The (betaalpha) (8) glycosidases: sequence and structure analyses suggest distant evolutionary relationships. Protein Eng 14:845–855
Ness JE, Welch M, Giver L, Bueno M, Cherry JR, Borchert TV, Stemmer WP, Minshull J (1999) DNA shuffling of subgenomic sequences of subtilisin. Nat Biotechnol 17:893–896
Nielsen JE, Beier L, Otzen D, Borchert TV, Frantzen HB, Andersen KV, Svendsen A (1999) Electrostatics in the active site of an α-amylase. Eur J Biochem 264:816–824
Nielsen JE, Borchert TV (2000) Protein engineering of bacterial α-amylase. Biochim Biophys Acta 1543:253–274
Nielsen JE, Borchert TV, Vriend G (2001) The determinants of α-amylase pH-activity profiles. Protein Eng 14:505–512
Peitsch MC (1996) ProMod and Swiss-Model: internet-based tools for automated comparative protein modelling. Biochem Soc Trans 24:274–279
Peitsch MC, Wells TN, Stampf DR, Sussman JL (1995) The Swiss-3DImage collection and PDB-browser on the World-Wide Web. Trends Biochem Sci 20:82–84
Peitsch MC, Herzyk P, Wells TN, Hubbard RE (1996) Automated modelling of the transmembrane region of G-protein coupled receptor by Swiss-model. Receptors Channels 4:161–164
Qian M, Haser R, Buisson G, Duee E, Payan F (1994) The active center of a mammalian α-amylase. Structure of the complex of a pancreatic α-amylase with a carbohydrate inhibitor refined to 2.2A resolution. Biochem 33:6284–6294
Richardson TB, Tan XQ, Frey G, Callen W, Cabell M, Lam D, Macomber J, Short JM, Robertson DE, Miller C (2002) A novel, high performance enzyme for starch liquefaction. J Biol Chem 277:26501–26507
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory, New York
Saito N (1973) A thermophilic extracellular α-amylase from Bacillus licheniformis. Arch Biochem Biophys 155:290–298
Schmidt-Dannert C, Arnold FH (1999) Directed evolution of industrial enzymes. Trends Biotechnol 17:135–136
Shaw A, Bott R (1996) Engineering enzymes for stability. Curr Opin Biotechnol 6:546–550
Shaw A, Bott R, Day AG (1999) Protein engineering of α-amylase for low pH performance. Curr Opin Biotechnol 10:349–352
Strokopytov B, Penninga D, Rozeboom HJ, Kalk KH, Dijkhuizen L, Dijkstra BW (1995) X-ray structure of cyclodextrin glycosyltransferase complexed with acarbose. Implications for the catalytic mechanism of glycosidases. Biochem 34:2234–2240
Svensson B, Sogaard M (1992) Protein engineering of amylases. Biochem Soc Trans 20:34–42
Svensson B (1994) Protein engineering in the α-amylase family: catalytic mechanism, substrate specificity and stability. Plant Mol Biol 25:141–157
Uitdehaag JC, Mosi R, Kalk KH, van der Veen BA, Dijkhuizen L, Withers SG, Dijkstra BW (1999) X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the α-amylase family. Nat Struct Biol 6:432–436
Violet M, Meunier JC (1989) Kinetic study of the irreversible thermal denaturation of Bacillus licheniformis α-amylase. Biochem J 263:665–670
Wong SL, Kawamura F, Doi RH (1986) Use of the Bacillus subtilis subtilisin signal peptide for efficient secretion of TEMβ-lactamase during growth. J Bacteriol 168:1005–1009
Wong SL (1995) Advances in the use of Bacillus subtilis for the expression and secretion of heterologous proteins. Curr Opin Biotechnol 6:517–522
Wu SC, Wong SL (1999) Development of improved pUB110-based vectors for expression and secretion studies in Bacillus subtilis. J Biochem 72:185–195
Wu XC, Lee W, Tran L, Wong SL (1991) Engineering a Bacillus subtilis expression–secretion system with a strain deficient in six extracellular proteases. J Bacteriol 173:4952–4558
Yasbin RE, Wilson GA, Young FE (1975) Transformation and transfection in lysogenic strains of Bacillus subtilis: evidence for selective induction of prophage. J Bacteriol 121:296–304
Yuuki T, Nomura T, Tezuka H, Tsuboi A, Yamagata H, Tsukagoshi N, Udaka S (1985) Complete nucleotide sequence of a gene coding for heat-and pH-stable α-amylase of Bacillus licheniformis: comparison of the amino acid sequences of three bacterial liquefying α-amylase deduced from the DNA sequences. J Biochem 98:1147–1156
Zhang XZ, Cui ZL, Hong Q, Li SP (2005) High-level expression and secretion of methyl parathion hydrolase in Bacillus subtilis WB800. Appl Environ Microbiol 71:4101–4103
Acknowledgments
The authors would like to thank Dr. Sui-Lam Wong for the supply of plasmid pWB980. Dr. Sui-Lam Wong is a senior medical scholar from University of Calgary. This work was supported by Key Program of Tianjin Science and Technology Development Plan (06YFGPSH03500).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Yh., Lu, Fp., Li, Y. et al. Characterisation of mutagenised acid-resistant alpha-amylase expressed in Bacillus subtilis WB600. Appl Microbiol Biotechnol 78, 85–94 (2008). https://doi.org/10.1007/s00253-007-1287-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-1287-z