
Abstract
The secretome of Penicillium funiculosum contains two family GH7 enzymes, one of which (designated XynA) has been described as a xylanase. This is unusual because it is the only xylanase in family GH7, which is mainly composed of cellobiohydrolases and endoglucanases, and also because XynA is highly similar to the cellobiohydrolase I from Talaromyces emersonii and Trichoderma reesei (72 and 65 % identity, respectively). To probe this enigma, we investigated the biochemical properties of XynA, notably its activity on xylans and β-d-glucans. A highly pure sample of XynA was obtained and used to perform hydrolysis tests on polysaccharides. These revealed that XynA is 100-fold more active on β-1,4-glucan than on xylan. Likewise, XynA was active on both 4-nitrophenyl-β-d-lactopyranoside (pNP-β-d-Lac) and 4-nitrophenyl-β-d-cellobioside (pNP-cellobiose), which shows that XynA is principally an exo-acting type 1 cellobiohydrolase enzyme that displays 5.2-fold higher performance on pNP-cellobiose than on pNP-β-d-Lac. Finally, analyses performed using cellodextrins as substrate revealed that XynA mainly produced cellobiose (C2) from substrates containing three or more glucosyl subunits, and that C2 inhibits XynA at high concentrations (IC50 C2 = 17.7 μM). Overall, this study revealed that XynA displays typical cellobiohydrolase 1 activity and confirms that the description of this enzyme in public databases should be definitively amended. Moreover, the data provided here complete the information provided by a previous proteomics investigation and reveal that P. funiculosum secretes a complete set of cellulose-degrading enzymes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant cell walls contain between 35 and 50 % of cellulose, a polymer composed of β-1,4 glucan chains that are organized into regular crystalline microfibrils (approximately 2.5 nm in diameter) that are embedded in a matrix of hemicellulose and lignin. To harness the glucose contained within cellulose, many microorganisms such as fungi and bacteria have devised enzymatic strategies to break down plant cell walls, thus overcoming the various obstacles that have evolved in plants to prevent this; For example, certain filamentous fungi produce potent secretomes that contain a wide variety of enzymes and accessory proteins that are able to penetrate plant cell walls and degrade cellulose microfibrils [8, 11]. Generally, cellulolytic enzyme systems contain three types of activity, which act cooperatively to bring about cellulose depolymerization: (1) endo-β-1,4-glucanase (EC 3.2.1.4) that randomly or specifically (depending on the enzyme) cleaves β-1,4-bonds between internal d-glucosyl units, (2) exo-β-1,4-glucanase or cellobiohydrolase (EC 3.2.1.91) that attacks from chain extremities to hydrolyze internal β-1,4-bonds and release cellodextrins, and (3) β-glucosidase (EC 3.2.1.21) that hydrolyzes cellodextrins, cleaving off glucose units from nonreducing ends [5, 9, 28]. In the CAZy classification of glycoside hydrolases (GH), such enzyme types are found in many families, but cellobiohydrolases (CBH) and many endo-β-1,4-glucanases (EG) are found in families GH5, 6, 7, 8, 9, 45, and 48 [15, 30].
Some CBHs and EGs are modular proteins, composed of a catalytic domain that is frequently, though not systematically, connected to a carbohydrate binding module (CBM) by a linker sequence that is rich in serine, threonine, and proline. These types of protein are made up of β-sheets arranged into β-sandwiches connected by loops [16]. Interestingly, CBHs and EGs possess quite similar three-dimensional (3D) structures that differ mainly in the loop regions. In CBHs, loops are generally longer and their amino acid sequence is generally different from that of EGs [10, 31]. Accordingly, the longer loops of CBHs close the substrate binding region, producing a tunnel-like configuration [2]. X-ray structure analysis of CBHs and EGs from family GH7 has revealed that the former possess approximately 11 substrate binding subsites, whereas EGs display 4–5 subsites, which form part of an open active site cleft. Overall, these differences partly explain the different mode of action of EGs and CBHs, the latter acting on β-1,4-linked glucan chains via a processive mechanism [30].
For many years, cellulases have been commercialized for a wide variety of industrial applications, which include textile modifications, detergent applications, and paper pulp refining [3]. However, one of the major focuses of research on cellulases has been the development of efficient cocktails for biorefining of lignocellulosic biomass, notably for bioethanol production [22, 33]. Similarly, the food and feed sector constitutes a considerable market for cellulase cocktails, which are used to increase the nutritional value and digestibility of cereals [1]. The main sources of industrial enzymes are microorganisms such as yeast and bacteria, although filamentous fungi are by far the most significant source, furnishing approximately 50 % of all industrial enzymes [6]. Although the filamentous fungus Trichoderma reesei is probably the most well-known producer of cellulolytic cocktails, other strains, such as Penicillium funiculosum, are also used to produce commercial cellulase cocktails that contain a wide variety of carbohydrate-acting enzymes, as revealed by an extensive proteomics analysis [13]. Among the enzymes found in the secretome of P. funiculosum (commercialized under the name Rovabio Excel by Adisseo S.A.S and previously by Rhodia Food UK Ltd.) are two proteins that belong to family GH7, one of which has been previously designated as XynA and biochemically described as a xylanase [12]. This is intriguing because XynA is the only known family GH7 xylanase so far described. Therefore, to follow up on the work performed by Furniss et al. [12], we now describe a thorough biochemical analysis of XynA, using the native enzyme purified directly from P. funiculosum secretome.
Materials and methods
Chemicals and reagents
Medium-viscosity barley β-glucan (MVβG), carboxymethyl cellulose—4 M (CMC), and wheat arabinoxylan (WAX) were purchased from Megazyme (Wicklow, Ireland), while birchwood xylan (BX), 4-nitrophenyl-β-d-lactopyranoside (pNP-β-d-Lac), and bovine serum albumin (BSA) were purchased from Sigma Aldrich (Buchs SG, Switzerland). 4-Nitrophenyl-β-d-cellobioside (pNP-cellobiose) was purchased from Carbosynth (Compton, UK).
Enzyme purification
The protein XynA was purified to near homogeneity in three steps using a sequence of hydrophobic interaction chromatography (HIC), then ion-exchange chromatography (IEC), and finally HIC once again, starting with the culture supernatant (1,700 mL) of P. funiculosum IMI 378536 supplied by Adisseo SAS (Anthony, France). The initial HIC was performed on a fast protein liquid chromatography (FPLC) system (AKTA Prime) equipped with a Phenyl Sepharose 6 FF High Sub column (GE Healthcare, France) equilibrated in 50 mM sodium phosphate buffer (pH 7), containing 1.2 M (NH4)2SO4. Proteins were eluted using a (NH4)2SO4 gradient (1.2 to 0 M over 10 column volumes at flow rate of 100 mL/min). Subsequently, the sample was applied to a Source 15 Q anion exchange column (GE Healthcare, France), equilibrated in 20 mM Tris–HCl, pH 7, and proteins were eluted using a two-step gradient (0–500 mM NaCl over 20 column volumes at 17 mL/min, then 500 mM to 1,000 mM NaCl over 10 column volumes at 17 mL/min). Finally, to polish the purified protein sample, fractions containing the protein of interest were pooled and dialyzed against 50 mM sodium phosphate buffer pH 7, and (NH4)2SO4 was added to final concentration of 1 M. The protein was injected onto a Phenyl FF 16/10 column (GE Healthcare, France), and then eluted using a (NH4)2SO4 gradient (1 to 0 M over 40 min). Fractions (5 mL) were collected, dialyzed against sodium phosphate buffer 50 mM, pH 7, and then assayed for β-xylanase (BX) and β-glucanase (MVβG) activities, respectively.
Protein sequence analysis
To verify that the purified sample was XynA, protein sequence analysis was performed. Briefly, the purified protein sample was first submitted to trypsin digestion, which was expected to generate fragments that would provide approximate protein sequence coverage of 50 %, and 5 μg of the sample was analyzed on a 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel running in TGS [25 mM Tris, 192 mM glycine, and 0.1 % (w/v) SDS, pH 8.3] buffer. Subsequently, nano liquid chromatography tandem mass spectrometry (LC–MS/MS) analysis was performed and the resulting sequences were used to interrogate the Sprot Tremb database.
Measurement of enzyme activity
Enzyme activity was assayed on polysaccharides, using the dinitrosalicylic acid (DNS) method to quantify reducing sugars in the reaction medium [23]. All reactions were performed for 20 min at 30 °C in 40 mM sodium citrate buffer, pH 5, containing BSA at 1 mg/mL, using MVβG (1 % w/v), CMC (2 % w/v), and BX (4 % w/v) or WAX (1 % w/v) to assay for β-glucanase and xylanase activity, respectively. At regular intervals during reactions, samples were removed, an equal volume of DNS reagent was added (100 μL), and the mixture was incubated for 20 min at 95 °C and then for 10 min on ice. Subsequently, 1 mL ultrapure water (room temperature) was added to each sample and the optical density (OD) was read at 540 nm using a Cary 100 Bio UV–Visible spectrophotometer (Agilent technologies, Santa Clara, USA). Activity was determined using standard curves prepared using xylose or glucose as appropriate. Activities were expressed as the quantity of glucose/cellobiose or xylose equivalents (μmol) released per min per liter and per mg of XynA. To determine the kinetic constants (K M and k cat), XynA was incubated with different concentrations of MVβG (0.1–1.4 % w/v) or CMC (0.1–1.6 % w/v). Afterwards, data were analyzed using weighted nonlinear least-squares regression and a one-site saturation algorithm in SigmaPlot software version 11.0 (SPSS, USA), which generated K M and k cat values. Accounting for the fact that it was not possible to obtain an accurate estimation of the molarity of the complex polysaccharides, values for K M were expressed as apparent values in units of mg mL−1.
To measure cellobiohydrolase activity, the chromogenic substrates pNP-β-d-Lac and pNP-cellobiose were employed. Reactions were conducted at 40 °C in 40 mM sodium citrate buffer, pH 5, and the release of pNP was monitored in a discontinuous manner by removing samples (40 μL) at regular intervals. The enzyme reaction was stopped by adding 200 μL 1 M sodium carbonate (pH 9), and the absorbance at 402 nm was measured using a Sunrise microplate reader (TECAN, Männedorf, Switzerland). The amount of free pNP was determined by comparing values against a standard curve prepared using a known range of pNP concentrations.
Temperature optimum and thermostability
To determine the optimum temperature for enzyme activity, reactions were performed using the conditions described above, but using different temperatures over the range 30–65 °C. Similarly, to determine thermostability, enzyme solutions in 40 mM sodium citrate buffer containing BSA (1 mg/mL), which was added as a carrier protein, pH 5, were incubated at temperatures in the range 35–50 °C, for 0, 15, 30, 60, 120, 180, 300, 480, and 1,440 min, before performing activity assays in standard conditions.
pH optimum and stability
To determine the optimum pH for enzyme activity, reactions were performed as described previously, but using different buffers (phosphate citrate buffer 40 mM, pH 2.5–3.5; sodium citrate buffer 40 mM, pH range 4–6; sodium phosphate buffer 40 mM, pH range 6.5–8). To determine the stability of the enzyme with regard to pH, before assaying the enzyme in standard conditions, the enzyme solution was incubated for 0, 15, 30, 60, 120, 180, 300, 480, and 1,440 min, in different pH conditions (2.5–8) using the different buffers mentioned above.
Determination of product profiles
To investigate the nature of the products generated by XynA acting on cellodextrins, reactions were performed using cellobiose (C2), cellotriose (C3), cellotetraose (C4), and cellopentaose (C5). To achieve this, XynA was incubated with solutions of these compounds (500 μM) in 40 mM sodium citrate buffer (pH 5), containing 1 mg/mL BSA. When C2 or C3 was used, the concentration of XynA was 37.5 μM, whereas this concentration was 0.375 μM when C4 or C5 was used. Reactions were started by enzyme addition, then aliquots were removed at regular intervals and immediately placed on ice. Subsequently, reactions were definitively arrested by enzyme inactivation at 95 °C for 5 min. Following centrifugation at 14,000 rpm for 10 min, the samples were diluted 10-fold in ultrapure water, filtered (Millex-GS, 0.22 μm, mixed cellulose esters, 25 mm, nonsterile; Millipore, USA) and injected onto an anion exchange Carbopac PA100 column (Dionex, Sunnyvale, CA, USA) running on a Dionex high-performance liquid chromatography (HPLC) system equipped with pulsed amperometric detection. Oligosaccharides were eluted in 50 mM NaOH using a linear gradient of sodium acetate (from 50 to 300 mM). Glucose, C2, C3, C4, and C5 were used as standards.
Determination of IC50 of cellobioses
To investigate inhibition of XynA by C2, hydrolysis of pNP-β-d-Lac by XynA in the presence of different concentrations (0, 10, 20, 200, and 500 μM) of C2 was performed. Reactions were conducted at 40 °C in 40 mM sodium citrate buffer, pH 5 containing 1 mg/mL BSA and 5 mM pNP-β-d-Lac. Release of pNP was monitored in a discontinuous manner by removing samples (40 μL) at regular intervals. The enzyme reaction was stopped by adding 200 μL 1 M sodium carbonate (pH 9), and the absorbance at 402 nm was measured using a Sunrise microplate spectrophotometer (TECAN, Männedorf, Switzerland). The amount of free pNP was determined by comparing values against a standard curve prepared using a known range of pNP concentrations. Afterward, the data were analyzed using a four-parameter logistic equation in SigmaPlot software (version 11.0) and the IC50 value (50 % inhibition concentration) was calculated. To estimate the K i value, the Cheng–Prusoff equation was used:
Results and discussion
In this study we set out to determine the exact specificity of XynA, a component of the secretome of P. funiculosum that has previously been described as a xylanase [12]. This is because XynA is classified in the GH7 family in the CAZy database and a BlastP search of the Protein Data Bank (PDB), using the sequence of XynA (Uniprot Q8WZJ4) as the query, reveals that the catalytic domain of XynA exhibits high similarity to that of family GH7 CBHs from T. emersonii (Uniprot Q8TFL9) and T. reesei (Uniprot P62694.1) (72 and 65 % identity, respectively). Moreover, the presence in XynA of a family 1 carbohydrate binding module (CBM) is consistent with the fact that this enzyme should possess CBH activity.
To provide unequivocal biochemical data, our initial intention was to clone and express the gene encoding XynA, but this proved to be impossible despite the fact that several expression hosts (bacteria and yeast) were tested. Therefore, to surmount this problem, we decided to devise a robust purification method that would yield a highly pure sample of XynA. Accordingly, a three-step purification method involving two HIC steps was implemented to purify XynA from Rovabio Excel™, mainly because XynA was not retained on the anion exchange column (second step), although this step was useful to remove some contaminating protein species. After the third step, XynA was obtained in a homogeneous, pure form that migrated to a position on Coomassie blue-stained SDS-PAGE (Fig. 1), corresponding to molecular mass of 52.5 kDa, which is the theoretical molecular weight for XynA.

SDS-PAGE analysis of the protein sample before purification (lane 1) and of the different fractions obtained using hydrophobic interaction chromatography (lanes 2–6). M is a protein molecular weight marker
To confirm that the majority protein species in the purified protein sample was XynA we performed protein sequence analysis, which generated sequence fragments that matched with the sequence of XynA (Uniprot Q8WZJ4), with fragments covering 34 % of the full protein sequence and a score of 19,692, compared with 91 for the sequence of the GH7 CBHA of Aspergillus niger (Uniprot Q9UVS9). This rather conclusive result confirmed that the purified species was XynA and thus allowed us to continue with the detailed analysis of the biochemical properties of the protein.
To begin the characterization of XynA, it was first necessary to determine which substrate would be most appropriate for further analyses. Therefore, activity screening was performed using substrates that would reveal both glucanase and xylanase activities. These experiments revealed that activity on MVβG was 100 times higher than the activities measured on BX (0.14 nmole equivalent xylose min−1 mg−1 of XynA) and WAX. These results contrast sharply with those published by Furniss et al. [12], which revealed activity of 45 μmol equivalent xylose min−1 mg−1 of XynA on BX.
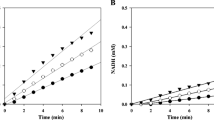
Having ascertained that MVβG is an appropriate substrate for XynA, it was possible to investigate some of the key physicochemical properties of XynA. The determination of thermoactivity and thermostability revealed that XynA is optimally active at approximately 55 °C (Fig. 2a), but at this temperature its thermostability would be low, since our data suggest that at 50 °C the enzyme’s half-life is less that 2.5 h (Fig. 2b). Likewise the determination of the optimum pH for activity indicated that XynA maintained >80 % activity on MVβG in the pH range 3–4.5 (Fig. 2c), which agrees reasonably well with the optimum pH range (3.7–5.2) reported by Furniss et al. [12]. Indeed, even after long periods of exposure (25 h) to acidic conditions (pH 2.5–5.5), the enzyme retained approximately 80 % of its original activity (Fig. 2d). Generally, the properties described here for XynA are highly similar to those of many other CBH enzymes from microbial sources; For example, CBH I from T. reesei displays optimal activity at approximately pH 4.8 and 50 °C [21].

Physicochemical properties of XynA. a Temperature dependency of XynA activity measured on medium-viscosity barley β-glucan. b Residual activity of XynA after incubation at 35 °C (filled circles), 40 °C (circles), 45 °C (filled inverted triangles), and 50 °C (triangles). c pH dependency of XynA activity measured on medium-viscosity barley β-glucan. d Residual activity of XynA after incubation at pH 2.5 (filled circles), pH 3 (circles), pH 3.5 (filled inverted triangles), pH 4 (triangles), pH 4.5 (filled squares), pH 5 (squares), and pH 5.5 (filled diamonds)
Regarding the kinetic parameters that were determined for the hydrolysis of the two β-glucans tested, the K M value for the hydrolysis of MVβG was markedly lower than that of the reaction involving CMC, and the catalytic constant for the former reaction was 18-fold higher, which is perfectly coherent with a cellobiohydrolase-like exoglucanase activity, rather than that of an endoglucanase (Table 1). Similarly, the use of the synthetic glycosides pNP-β-d-Lac and pNP-cellobiose clearly revealed that XynA is capable of exo-acting activity, and significantly, the catalytic constant for hydrolysis of pNP-cellobiose was 5.2-fold higher than for pNP-β-d-Lac, thus revealing clear CBH I character. It is also noteworthy that the K M value displayed by XynA for hydrolysis of pNP-cellobiose (0.238 mM) is quite similar to those of other CBHs whose K M values are often in the range 0.1–0.8 mM [4, 18, 20, 26]. Likewise, the turnover numbers measured on pNP-cellobiose and pNP-β-d-Lac were remarkably high, since the k cat value measured for Cel7A of T. reesei or Cel7D of Phanerochaete chrysosporium on pNP-β-d-Lac was only 0.093 and 0.17 s−1, respectively, and the k cat value measured for Cex of Cellulomonas fimi on pNP-cellobiose was 15.8 s−1 [24, 25].
Using different cellodextrins containing two to five glucosyl units as substrates revealed that XynA was active on all of these, although cellobiose was a poor substrate, being very slowly hydrolyzed to glucose (Fig. 3a–d). Indeed, after 24 h, the concentration of cellobiose had only diminished from 350 to 260 μM (Fig. 3d). In contrast, C3 was rapidly hydrolyzed to C2 and glucose, with the reaction reaching near completion after only 10 min (Fig. 3a). A highly similar, rapid reaction was observed for C4, with mainly C2 being produced, accompanied by only small amounts of glucose and C3 (Fig. 3b). Finally, it is noteworthy that C2 was also the major product of C5 hydrolysis, though C3 and glucose are produced, indicating that the initial hydrolysis of C5 produces C2 and C3, and that subsequent hydrolysis of C3 produces C2 and glucose (Fig. 3c). Overall, this behavior is similar to that described for Cel7A of T. reesei, although this latter does not release any trace of glucose when hydrolyzing cellodextrins [14]. Interestingly, in the case of C5, the use of higher concentrations of substrate >50 μM revealed an increasingly obvious inhibitory effect, which became severe when concentrations >100 μM were used. Since it is well known that CBHs are often inhibited by cellobiose [17], hydrolysis of pNP-β-d-Lac in the presence of increasing concentrations of cellobiose was investigated. This analysis provided an IC50 (C2) value of 17.7 μM. Likewise, using the Cheng–Prusoff equation, it was possible to generate a K i value of 1.8 μM [7]. Overall, these results are consistent with the observed inhibition of XynA, and the IC50 value measured for the T. reesei CBH I (approximately 35.3 μM), using MeUmbG2 as the substrate [32]. Nevertheless, the estimated value of K i is lower than that observed for the P. chrysosporium CBH I (K i C2 = 65 μM) acting on pNP-cellobiose [19]. Considering that some CBH1 enzymes have been reported to operate normally in the presence of C2 up to concentrations of 0.5–2 mM, it is possible conclude that XynA is quite sensitive to inhibition [27, 29].

XynA-mediated hydrolysis of cellotriose (a), cellotetraose (b), cellopentaose (c), and cellobiose (d). Symbols are cellotriose (filled inverted triangles), cellobiose (circles), glucose (filled circles), cellotetraose (filled triangles), and cellopentaose (filled squares)
Conclusions
In this study we set out to provide new biochemical data for XynA of P. funiculosum, an enzyme that is a member of the CAZy family GH7 and that has been previously described as a xylanase, despite the fact that no other xylanases have so far been identified in family GH7. In total contrast to the data presented by Furniss et al. [12], we failed to detect any significant xylanase activity, but instead clearly measured activities on β-d-glucans and cellodextrins, with catalytic behavior that corresponds to that of a type 1 cellobiohydrolase or CBH I. Therefore, based on our data, we propose that the description of XynA contained within the CAZy database and elsewhere should be amended and that the enzyme should be renamed Cel7A. Beyond these recommendations, we note that, based on our data and on those already published by Guais et al. [13], P. funiculosum is equipped with a full arsenal of cellulose-degrading enzymes and could thus be an interesting candidate for the production of a lignocellulosic biomass-degrading enzyme cocktail.
References
Ahuja SK, Ferreira G, Moreira A (2004) Utilization of enzymes for environmental applications. Crit Rev Biotechnol 24:125–154. doi:10.1080/07388550490493726
Beckham GT, Bomble YJ, Bayer E, Himmel ME, Crowley MF (2011) Applications of computational science for understanding enzymatic deconstruction of cellulose. Curr Opin Biotechnol 22:231–238. doi:10.1016/j.copbio.2010.11.005
Bhat MK (2000) Cellulases and related enzymes in biotechnology. Biotechnol Adv 18:355–383. doi:10.1016/S0734-9750(00)00041-0
Bhat S (2001) Isolation and characterisation of a major cellobiohydrolase (S8) and a major endoglucanase (S11) subunit from the cellulosome of Clostridium thermocellum. Anaerobe 7:171–179. doi:10.1006/anae.2001.0374
Boisset C, Fraschini C, Schülein M, Chanzy H, Henrissat B, Schu M (2000) Imaging the enzymatic digestion of bacterial cellulose ribbons reveals the endo character of the cellobiohydrolase Cel6A from Humicola insolens and its mode of synergy with cellobiohydrolase Cel7A. Appl Environ Microbiol 66:1444–1452. doi:10.1128/AEM.66.4.1444-1452.2000
Boopathy R (1994) Enzyme technology in food and health industries. Indian Food Ind 13:22–31
Cheng Y, Prusoff W (1973) Relationship between the inhibition constant (K i ) and the concentration of inhibitor which causes 50 per cent inhibition (I 50 ) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108. doi:10.1016/0006-2952(73)90196-2
Couturier M, Haon M, Coutinho PM, Henrissat B, Lesage-Meessen L, Berrin JG (2011) Podospora anserina hemicellulases potentiate the Trichoderma reesei secretome for saccharification of lignocellulosic biomass. Appl Environ Microbiol 77:237–246. doi:10.1128/AEM.01761-10
Davies G, Henrissat B (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3:853–859. doi:10.1016/S0969-2126(01)00220-9
Divne C, Ståhlberg J, Teeri T, Jones T (1998) High-resolution crystal structures reveal how a cellulose chain is bound in the 50 Å long tunnel of cellobiohydrolase I from Trichoderma reesei. J Mol Biol 275:309–325. doi:10.1006/jmbi.1997.1437
Eriksson KEL, Blanchette RA, Ander P (1990) Microbial and enzymatic degradation of wood and wood components. In: Microbial and enzymatic degradation of wood and wood components. Springer, New York
Furniss CS, Williamson G, Kroon PA (2005) The substrate specificity and susceptibility to wheat inhibitor proteins of Penicillium funiculosum xylanases from a commercial enzyme preparation. J Sci Food Agric 85:574–582. doi:10.1002/jsfa.1984
Guais O, Borderies G, Pichereaux C, Maestracci M, Neugnot V, Rossignol M, François JM (2008) Proteomics analysis of “Rovabio™ Excel”, a secreted protein cocktail from the filamentous fungus Penicillium funiculosum grown under industrial process fermentation. J Ind Microbiol Biotechnol 35:1659–1668. doi:10.1007/s10295-008-0430-x
Harjunpää V, Teleman A, Koivulaa A, Ruohonen L, Teeri TT, Telemano O, Drakenberg T (1996) Cello-oligosaccharide hydrolysis by cellobiohydrolase II from Trichoderma reesei. Association and rate constants derived from an analysis of progress curves. Eur J Biochem 240:584–591
Henrissat B (1991) A classification of glycosyl hydrolases based on amino-acid sequence similarities. Biochem J 280:309–316
Henrissat B, Davies G (1997) Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol 7:637–644. doi:10.1016/S0959-440X(97)80072-3
Holtzapple M, Cognata M, Shu Y, Hendrickson C (1990) Inhibition of Trichoderma reesei cellulase by sugars and solvents. Biotechnol Bioeng 36:275–287. doi:10.1002/bit.260360310
Huang L, Forsberg CW (1988) Purification and comparison of the periplasmic and extracellular forms of the cellodextrinase from Bacteroides succinogenes. Appl Environ Microbiol 54:1488–1493
Igarashi K, Samejima M, Eriksson KE (1998) Cellobiose dehydrogenase enhances Phanerochaete chrysosporium cellobiohydrolase I activity by relieving product inhibition. Eur J Biochem 253:101–106
Lee KM (2011) Characterization of cellobiohydrolase from a newly isolated strain of Agaricus arvencis. J Microbiol Biotechnol 21:711–718. doi:10.4014/jmb.1102.02001
Medve J, Lee D, Tjerneld F (1998) Ion-exchange chromatographic purification and quantitative analysis of Trichoderma reesei cellulases cellobiohydrolase I, II and endoglucanase II by fast protein liquid chromatography. Technology 808:153–165. doi:10.1016/S0021-9673(98)00132-0
Merino ST, Cherry J (2007) Progress and challenges in enzyme development for Biomass utilization. Biofuels 108:95–120. doi:10.1007/10_2007_066
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428. doi:10.1021/ac60147a030
Muños IG, Ubhayasekera W, Henriksson H, Szabo I, Pettersson G, Johansson G, Mowbray SL, Ståhlberg J (2001) Family 7 cellobiohydrolases from Phanerochaete chrysosporium : crystal structure of the catalytic module of Cel7D (CBH58) at 1.32 Å resolution and homology models of the isozymes. J Molecular Biol 314:1097–1111. doi:10.1006/jmbi.2001.5180
von Ossowski I, Ståhlberg J, Koivula A, Piens K, Becker D, Boer H, Harle R, Harris M, Divne C, Mahdi S, Zhao Y, Driguez H, Claeyssens M, Sinnott ML, Teeri TT (2003) Engineering the exo-loop of Trichoderma reesei cellobiohydrolase, Cel7A. A comparison with Phanerochaete chrysosporium Cel7D. J Mol Biol 333:817–829. doi:10.1016/S0022-2836(03)00881-7
Piens K, Ståhlberg J, Nerinckx W, Teeri TT, Claeyssens M (2004) Structure-reactivity studies of Trichoderma reesei cellobiohydrolase Cel7A. In: Lignocellulose biodegradation, 12: 207–226. Doi:10.1021/bk-2004-0889.ch012
Schmidhalter DR, Canevascini G (1993) Purification and characterisation of 2 exocellobiohydrolases from the brown-rot fungus Coniophora puteana (schum ex-fr) Karst. Arch Biochem Biophys 300:551–558. doi:10.1006/abbi.1993.1076
Schou C, Rasmussen G, Kaltoft M, Henrissat B, Schulein M (1993) Stereochimie, specificity and kinetics of the hydrolysis of reduced cellodextrins by 9 cellulases. Eur J Biochem 217:947–953. doi:10.1111/j.1432-1033.1993.tb18325.x
Schülein M (1997) Enzymatic properties of cellulases from Humicola insolens. J Biotechnol 57:71–81. doi:10.1016/S0168-1656(97)00090-4
Schülein M (2000) Protein engineering of cellulases. Biochim Biophys Acta 1543:239–252. doi:10.1016/S0167-4838(00)00247-8
Sulzenbacher G, Schülein M, Davies G (1997) Structure of the endoglucanase I from Fusarium oxysporum: native, cellobiose, and 3,4-epoxybutyl beta-d-cellobioside-inhibited forms, at 2.3 A resolution. Biochemistry 36:5902–5911. doi:10.1021/bi962963
van Tilbeurgh H, Bhikhabhai R, Pettersson LG, Claeyssens M (1984) Separation of endo- and exo-type cellulases using a new affinity chromatography method. FEBS Lett 169:215–218. doi:10.1016/0014-5793(84)80321-X
Wilson DB (2009) Cellulases and biofuels. Curr Opin Biotechnol 20:295–299. doi:10.1016/j.copbio.2009.05.007
Acknowledgments
The authors would like to thank the Agence National de la Recherche et de la Technologie for financial support accorded to Hélène Texier. Dr. Monsarrat and his team at the IPBS (Institut de Pharmacologie et de Biologie Struturale, Toulouse, France) are thanked for the protein sequence and glycosylation analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Texier, H., Dumon, C., Neugnot-Roux, V. et al. Redefining XynA from Penicillium funiculosum IMI 378536 as a GH7 cellobiohydrolase. J Ind Microbiol Biotechnol 39, 1569–1576 (2012). https://doi.org/10.1007/s10295-012-1166-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-012-1166-1