
Abstract
The thermostability of xylanase XynR8 from uncultured Neocallimastigales rumen fungal was improved by combining random point mutagenesis with site-directed mutagenesis guided by rational design, and a thermostable variant, XynR8_VNE, was identified. This variant contained three amino acid substitutions, I38V, D137N and G151E, and showed an increased melting temperature of 8.8 °C in comparison with the wild type. At 65 °C the wild-type enzyme lost all of its activity after treatment for 30 min, but XynR8_VNE retained about 65 % activity. To elucidate the mechanism of thermal stabilization, three-dimensional structures were predicted for XynR8 and its variant. We found that the tight packing density and new salt bridge caused by the substitutions may be responsible for the improved thermostability. These three substitutions are located in the N-terminus, cord and α-helix domains, respectively. Hence, the stability of these three domains may be crucial for the thermostability of family 11 xylanases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Xylanases are grouped into two families, F and G, which are also known as glycosyl hydrolase (GH) families 10 and 11, respectively [7], based on sequence homology and hydrophobic cluster analysis. The latter xylanases are typical hemicellulases, and are widely used in food, feed, and paper and pulp industries [3]. The GH11 xylanases derived from eukaryotic and bacterial species share 40–90 % sequence identities [22, 23]. The three-dimensional structure of a GH11 xylanase usually comprises two β-sheets and a single three-turn α-helix, which resemble a partly closed right hand [20] and may divide into four domains: thumb, palm, fingers and cord, respectively [5, 6, 11, 23].
Thermostable xylanases are widely used in many biotechnological processes operated at high temperatures, such as in the petroleum industry, paper and pulp industry [26]. Sequence alignments, mutagenesis studies and crystal structure analyses have shown that the enhanced thermostability of thermophilic xylanases may be attributed to an array of minor differences, when compared to mesophilic xylanases of the same family. These differences include more charged surface residues [24], more salt bridges and hydrogen bonds [6, 30], an improved internal packing [6, 33], the presence of thermostabilizing domains [28], and the introduction of disulphide bridges at the N- or C-termini or in the α-helix domain [31, 32]. The reported studies have suggested that the N-terminus and α-helix domain of the xylanase are critical for its thermostability [4, 19, 21, 31, 32, 34]. However, this information is still not enough to guide the improvement of the xylanase thermostability.
Site-directed mutagenesis [4, 9, 14, 35] and directed evolution [8, 16, 27, 35] are two major ways to improve the thermostability of enzymes. In this study, a GH11 xylanase xynR8, which was obtained directly from a mixed DNA sample prepared from unpurified rumen fungal cultures, was utilized to screen thermostable mutants by directed evolution and site-directed mutagenesis [13]. The full coding sequence of the wild-type gene xynR8 is 885 bp in length and encodes a 295-residue protein. XynR8 belongs to the endoxylanases, because the main hydrolysis products of it are xylobiose, xylotriose and xylotetrose [13]. Finally, we obtained a thermostable mutant of xynR8, and further analysis indicated that an improved packing density and the formation of a new salt bridge in this mutant may be responsible for the improved thermostability. According to the location of the point mutations, it seems that, in addition to the N-terminus and the α-helix domains, the cord domain of the GH11 xylanase may also be important for its thermostability. Thus, the stability of these three domains may contribute to the thermostability of GH11 xylanases.
Materials and methods
Analysis of the xynR8 core domain
The amino acid sequence of XynR8 was analysed using the National Center for Biotechnology Information Conserved Domain Database (NCBI CDD; http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) [15] and the Simple Modular Architecture Research Tool) (SMART; http://smart.embl-heidelberg.de) [12], and then the core domain of xylanase xynR8 was identified (200 residues in length, from 30 aa to 229 aa). xynR8 and its truncated derivative xynR8_CS (222 residues in length, from 16 aa to 237 aa) were cloned into pET21a (Novagen, Darmstadt, Germany), and the resulting reconstructed plasmids were designated pET21a/xynR8 and pET21a/xynR8_CS, respectively.
Recombinant proteins expression and purification
The reconstructed plasmids were transformed into Escherichia coli (E. coli) BL21 (DE3) (Invitrogen, Carlsbad, California) for protein expression. Protein purification was carried out by utilizing Ni–NTA resin (Novagen). The purity of final proteins was better than 95 %, and was used to analyse profiles of enzymatic activities and kinetic parameters. Against the reference of bovine serum albumin, the concentrations of the purified proteins were determined using the BCA protein assay kit (Thermo Fisher Scientific, Inc.).
Constructing the xylanase variants library by error-prone PCR
To generate a xynR8_CS variants library, error-prone PCR was carried out using the GeneMorph II random mutagenesis kit (Stratagene, Santa Clara, California) with the plasmid pET21a/xynR8_CS as the template. The reaction mixture contained 1.25 units of Mutazyme II DNA polymerase, 2.5 μL of 10× Mutazyme II reaction buffer, 0.8 mM dNTP mix, 0.25 μM concentrations each of flanking primer, 1 ng of pET21a/xynR8_CS plasmid and 20.75 μL water in a total volume of 25 μL. The mixture was heated at 94 °C for 30 s, followed by 30 cycles of incubation at 94 °C for 30 s, 57 °C for 40 s and 72 °C for 45 s. The PCR products were cloned into pET21a vector and then transformed into competent E. coli BL21 (DE3). All primers used in this research are listed in Table 1.
Library screening of the thermostable variants
Library screening was conducted on 96-well plates [18]. E. coli BL21 (DE3) transformants were picked with sterile toothpicks separately, and incubated in 96-well plates containing 200 μL of LB supplemented with 0.1 mM IPTG and 100 μg/mL ampicillin in each well. The cultures were incubated at 22 °C with shaking at 220 rpm until the OD600 reached about 1.0. The bacterial cells were harvested by centrifuging at 4,000 rpm for 10 min, and then resuspended in 150 μL of 50 mM Na2HPO4–NaH2PO4 buffer (pH 7.2). The cells were lysed by freezing the 96-well plates at −80 °C for 10 min, and then thawing the plates quickly at 37 °C for 10 min, and these processes were performed three times. The cell extracts from the wild-type xynR8_CS and the clones of mutant library were all preheated at 75 °C for 5 min, and incubated with substrates at 58 °C for 10 min to assay the reduced activities by the 3,5-dinitrosalicylic acid (DNS) method as below [18, 33]. The mutants that exhibited higher reduced activities than the wild type were identified. They were also double checked, by sequencing and culturing in 7 mL medium with the same treatment for further enzymatic activity assays.
Enzymatic activity assays
Enzymatic activities of xylanases were determined in 200 μL reaction mixture containing appropriate enzymes, 1 % (w/v) brichwood xylan (Sigma, Steinheim, Germany) as the substrate and 50 mM Na2HPO4–NaH2PO4 buffer. The DNS method was used for enzymatic activity determination [17] with d-xylose as the reference. One unit of enzymatic activity (U) was defined as 1 μmol sugar released per minute.
The temperature optima were determined by incubating the enzymes and substrates in 50 mM Na2HPO4–NaH2PO4 buffer at pH 7.2 for 10 min, at an interval of 5 °C from 45 to 80 °C [4]. The optimal pH of xylanase was measured at the optimal temperature of enzymes for 10 min from pH 4.0 to 8.0, with 50 mM citric acid buffer covering a pH range of 4.0 to 6.0 and 50 mM Na2HPO4–NaH2PO4 buffer covering a pH range of 5.5 to 8.0 [16].
The thermostability, also called the time-dependent activity, was measured in the presence or absence of the substrate [4]. In the presence of the substrate, purified enzymes were incubated with 1 % birchwood xylan at 65 °C and pH 7.2 for 60 min, and samples were withdrawn every 10 min after incubation. The accumulated product during the first 10 min of hydrolysis was set to 100 %. The relationship between accumulated product of reduced sugar and reaction time was determined. In the absence of the substrate, the residual activity was measured after preheating the enzyme at 65 °C and pH 7.2 for 0–60 min. The natural logarithm of the residual activity is a linear function with the inactivation time: ln(residual activity) = −k i t, where k i is the inactivation rate and t is the inactivation time [26]. The half-life (t 1/2) was calculated from k i (t 1/2 = ln2/k i). All thermostability results are reported as the mean values of triplicate measurements.
Reduced sugar was measured after incubating each enzyme with the substrate birchwood xylan at the optimal temperature and pH 7.2 for 10 min, and the Michaelis–Menten constants (K m and k cat) and activation energy (E a) were calculated as suggested previously [26]. The substrate concentrations were varied from 1 to 10 μg/μL typically.
Circular dichroism assays
Circular dichroism data of xylanase XynR8 were collected on a JASCO 175 spectropolarimeter. Far UV–CD wavelength scans (between 200 and 250 nm) at different protein concentration (0.01–0.15 mg mL−1) in 1 mM Na2HPO4–NaH2PO4 buffer (pH 7.2), and temperatures (30–85 °C) at 1 °C min−1, were collected in a 1-cm-path-length cuvette. The sample was cooled after being denatured to measure the reversibility of the protein denaturation at the rate of 1 °C min−1. Integration time was set to 5 min for data acquisition, from which the buffer blank spectrum was subtracted. When the spectra show defined minima, the melting temperature (T m, the midpoint of protein unfolding transition) was determined by extracting the ellipticity values of the enzymes at the respective wavelength [29].
Modelling of three-dimensional structures and protein design
Swiss-Model (http://swissmodel.expasy.org/) was utilized to model the three-dimensional structure of XynR8_CS and its mutants [1]. The template structure is 2c1fA (PDB code), which shares the highest sequence identity (90.8 %) with XynR8_CS among the proteins in the Protein Data Bank (PDB) (http://www.rcsb.org/). RosettaDesign software can identify low free energy sequences for target protein backbones and also stabilize proteins or create new protein structures [14]. By keeping other residues in the sequence as the native amino acids, and substituting the given sites 38, 137 and 151 with 20 allowed amino acids, RosettaDesign predicted the amino acids at sites 38, 137 and 151 that lead to low free energies. At least one thousand lowest energy sequences were predicted for each site, and the most commonly occurring residues at the given sites were chosen.
Results
Enzymatic characters of XynR8 and XynR8_CS
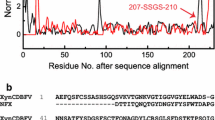
Considering that it would be easier for a xylanase with small molecular weight to enter the centre of hemicelluloses for enzymatic degradation than a bigger one [10], we cloned a truncated fragment of xynR8 (16–237 aa). It contained the core domain of GH11 and was designated xynR8_CS (Fig. 1). XynR8 and XynR8_CS were then separately expressed in E. coli, and their enzymatic properties were analysed. As shown in Fig. 1 and Table 2, the optimal pH values of XynR8 and XynR8_CS were pH 6.5 and pH 7.2, respectively. The optimal temperature was 55 °C for XynR8 and 58 °C for XynR8_CS (Fig. 1, Table 2). Accordingly, we concluded that the core domain of XynR8 retained the catalytic properties of the full-length XynR8. The truncated derivative XynR8_CS was more thermostable and alkali-stable than XynR8, and thus suitable for further studies.

Schematic diagram of XynR8_CS and enzymatic characters of XynR8 and its mutants. a Schematic diagram of the amino acid sequence region of XynR8 and XynR8_CS. The conserved domain of GH11 xylanase was predicted by utilizing CDD and SMART. b Thermostability and thermophilicity of XynR8 and its mutants. Cell lysates were incubated at elevated temperatures for 10 min in the absence of substrate and the relative activities were determined. c pH-dependence profiles of activities of XynR8 and its mutants. Purified enzymes were incubated at pH 4.0–8.0 over 10 min at their optimal temperature for XynR8 and its mutants
Mutations at I38, D137 and G151 improved the thermostablility of XynR8
To obtain thermostable mutants, the gene xynR8_CS was amplified using error-prone PCR technique with Taq polymerase, under conditions that promote the mis-incorporation of nucleotides. About 2,200 clones were all screened for their thermostability in 96-well plates, and three clones, 1F8, 5G9 and 17G6, which exhibited higher thermostability than xynR8_CS, were identified. As shown in Fig. 2, the mutant 1F8 contains a point mutation D137G, the mutant 5G9 contains a point mutation I38V, and the mutant 17G6 contains three mutations: A104T, F116L, and G151D.

Lineage of thermostable mutants of xylanase XynR8. Amino acid substitutions accumulated through random point mutagenesis, site-directed mutagenesis, RosettaDesign prediction and recombination are shown
To elucidate the effects of residue substitution at sites 38, 104, 116, 137 and 151 on the thermostability of XynR8, three mutants which contained A104T, F116L and G151D, separately, were constructed by directed mutagenesis (Fig. 2). Including the mutants I38V and D137G, all of them were expressed in E. coli and purified to determine their half-lives at 65 °C (Table 3). The half-lives of the mutants I38V, D137G and G151D were found to be longer than that of XynR8_CS. On the contrary, the half-lives of the mutants A104T and F116L were shorter than that of XynR8_CS (Table 3).
To evaluate the combinational effects of the five sites on the enzyme stability, we constructed a series of mutants which contained a combination of mutations at different sites (Fig. 2), and their half-life times at 65 °C were compared. These mutants were designated 1F8B to 1F8E (Fig. 2). As shown in Table 3, their half-life differences suggested that mutations Val-38, Gly-137 and Asp-151 might contribute to the increased thermostability of XynR8, and the mutations Thr-104 and Leu-116 decreased the enzymatic thermostability. These results were consistent with those of single locus mutations as shown above. Hence, we concluded that the mutations of Val-38, Gly-137 and Asp-151 could increase the thermostability of XynR8, but Thr-104 and Leu-116 may decrease its stability. On the basis of this conclusion, a mutant containing all three point mutations, I38V, D137G and G151D, was constructed finally and designated XynR8_VGD (Fig. 2).
Locations of three mutation sites on the three-dimensional structure of xylanase XynR8
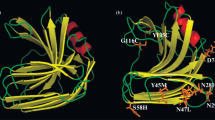
In order to explore the possible thermostability mechanism of the mutant, three-dimensional (3D) structures of XynR8_CS and XynR8_VGD were constructed, by utilizing a homology modelling approach via the Swiss-Model webserver (http://swissmodel.expasy.org). XynR8_CS has 90.8 % amino acid sequence identity with the modelling template, the GH11 xylanase NpXyn11A from Neocallimastix patriciarum [25]. As shown in Fig. 3a, the 3D structure of XynR8_CS consisted of one short α-helix and 16 β-strands, and the β-strand B6 was divided into B6a and B6b. Among them, the β-strands B6a and B9 to B13 were involved in interaction with the α-helix, and formed an α-helix domain. Residue Ile-38 was located in the β-strand B3 (Fig. 3a, b), and it was also found that both residues Asp-137 and Gly-151 were amino acids on the surface and were located in the loops of XynR8_CS (Fig. 3a, d–i).

Location of three mutations in 3D structures of XynR8. a Xylanase XynR8_CS comprises two β-sheets (16 β-strands) and an α-helix forming a so-called β-sandwich structure. The different parts of the enzyme are named on the basis of the right hand, containing fingers domain, thumb domain, palm domain, cord domain and α-helix domain. b The location and surrounding residues of site I38, which were L18, V20, H27, G47, M49, F70, A72. c The location of mutant V38. d The location and surrounding residues of site D137, which were W135, V136, W138, V139 and Q100. e The location of mutant G137. f The location of mutant N137. g The location and surrounding residue of site G151, which was R184. h The location of mutant D151. i The location of mutant E151. This figure and subsequent structural representations were drawn with PyMol (http://pymol.sourceforge.net/)
Selective mutagenesis at sites 38, 137 and 151 based on RosettaDesign analysis
To further identify the thermostability mechanisms of the mutation sites, the program RosettaDesign was utilized to computationally screen the most compatible one from all 20 allowed amino acids at the three sites (38, 137 and 151), and predict the most commonly occurring (i.e. stable) amino acids at these sites. On the basis of the predicted results listed in Supplementary Table S1, site-directed mutagenesis was carried out and these three sites were substituted by four or five other amino acids (i.e. three or four most commonly occurring substitutions, and one least commonly occurring substitution as the negative control) to construct mutants (Table 3). The half-lives of XynR8_CS and all these mutants were analysed. As shown in Table 3, the mutants I38V, D137N and G151E were the best substitutions at sites 38, 137 and 151, respectively, among all the constructed mutants. The mutants I38F and G151P lose their activities in the hydrolysis of xylans. Eventually, a mutant containing I38V, D137N and G151E was constructed on the basis of these results, and it was designated XynR8_VNE (Fig. 2). The 3D structure of XynR8_VNE was also predicted by the Swiss-Model webserver.
Activity profiles of XynR8_CS, XynR8_VGD and XynR8_VNE
The kinetic parameters K m, k cat, T m and E a (activation energy) of XynR8_CS and the mutants were also measured. The K m value of the wild-type XynR8_CS was 25.2 ± 0.9 mg mL−1; the mutants XynR8_VGD and XynR8_VNE displayed a slight decrease in the substrate affinity, and their K m values were 31.5 ± 1.4 mg mL−1 and 28.3 ± 1.1 mg mL−1, respectively (Table 2). However, the k cat values of XynR8_VGD and XynR8_VNE were 104.0 ± 5.2 s−1 and 91.8 ± 3.7 s−1, respectively, showing a remarkable increase when compared with XynR8_CS (k cat 22.5 ± 1.6 s−1). Therefore, both mutants possessed increased catalytic efficiency, as suggested by the k cat K −1m ratio, which was about fourfold the ratio of XynR8_CS (Table 2).
The optimal temperatures of XynR8_CS, XynR8_VGD and XynR8_VNE were 58, 65 and 68 °C, respectively, and the pH optima of those three enzymes were similar (Table 2). The T m values of XynR8_VGD and XynR8_VNE were 6.3 and 8.8 °C higher than that of XynR8_CS, respectively (Table 2). Their half-lives at 65 °C were 27.8, 62.7 and 7.4 min, respectively. XynR8_CS lost all of its activity after being treated at 65 °C for 30 min. However, XynR8_VGD and XynR8_VNE retained about 40 and 65 % activity, respectively, under the same treatment (Fig. 4a). In the presence of the substrates, the amounts of reaction products of XynR8_VGD and XynR8_VNE for a reaction time of 60 min were about 153 and 193 %, respectively; however, XynR8_CS released only about 124 % reducing sugar under the same treatment (Fig. 4b). These results confirmed that both mutants possessed higher thermostability than XynR8_CS.

Time-dependence profiles of XynR8_CS, XynR8_VGD and XynR8_VNE at 65 °C and pH 7.2. Effect of temperature on the stability of XynR8_CS and its mutants progeny at 65 °C in the absence (a) or presence of substrates (b). Samples were withdrawn every 10 min after incubation and the amount of released reducing sugar was measured by DNS method as described. To compare different enzymes, the activity detected after 10 min enzyme reaction was set as 100 % (b)
Discussion
In this study, we obtained a thermostable variant of XynR8, XynR8_VNE. It contains three amino acid substitutions: I38V, D137N and G151E, which are located at the N-terminus, cord domain and α-helix domain, respectively. XynR8_VNE showed an increase in melting temperature of 8.8 °C when compared with XynR8_CS, without compromising its specific activity. At 65 °C the wild-type enzyme lost all of its activity after treatment at 65 °C for 30 min, but the XynR8_VNE retained about 65 % activity.
According to the modelling results, it was found that site Ile-38 was located at the N-terminus β-strand B3 and surrounded by six hydrophobic amino acids and one hydrophilic amino acid (Fig. 3b), forming a hydrophobic core. Both Ile and Val are hydrophobic amino acids and share the most similar configuration and characteristics; however, the side chain of Val contains one less methyl than that of Ile, which resulted in the configuration of Val being smaller and more symmetrical. When Ile-38 was replaced by Val-38, the surrounding amino acids Leu-18, Val-20, Gly-47 and Met-49 could pack much more tightly than before, which led to an increase of the packing density at the N-terminus. Considering that the mutant I38V (Fig. 3c) was more thermostable than XynR8_CS, it seemed that the increased packing density of the enzyme was responsible for its improved thermostability. Zhang et al. also found the replacement of Ile by Val improved the thermostability of xylanase XT6 [35], which suggested that the substitution of Ile with Val in a hydrophobic core region could increase the enzyme stability. Previous studies showed that the mutations in the N-terminus of the GH11 xylanases improved their thermostability, and we found almost all of these mutations located at β-strands B1–B6a [4, 19, 21, 31, 32, 34]. Unlike the C-terminus of GH11 xylanase, which is buried in the centre of β-sheets (Fig. 3a), the N-terminus of GH11 xylanase is located in the edge of β-sheets and exposed to the hydrophilic environment (Fig. 3a), which means the N-terminus is more easily denatured than the C-terminus at high temperature. Therefore, we concluded that the N-terminus (β-strands B1–B6a) of GH11 xylanase could be very important for their stability, and improvement of the N-terminus packing density has the possibility to enhance the thermostability of GH11 xylanase.
The Asp-137 was located in the loop between β-strands B9 and B10 of XynR8, which was designated the cord domain (Fig. 3a, d). All residues at the cord domain are almost exposed to the hydrophilic environment, which may cause this domain to be quite frangible. The Asp-137 was surrounded by four hydrophobic amino acids (Trp-135, Val-136, Trp-138 and Val-139) and close to the residue Gln-100, which was located at β-strand B7. This hydrophobic patch could stabilize the 3D structure of the enzyme by the aromatic interaction between Trp and Val [4]. However, the presence of the hydrophilic amino acid Asp-137 in this patch might destroy the hydrophobic interaction and decrease the enzyme’s stability. Hence, the substitution of Asp-137 with Gly-137 could greatly reduce the destructive effect because of the small configuration of Gly, and the enzyme’s thermostability improved.
On the other hand, an attractive interaction was also found between Asp-137 and Gln-100 because of their characteristics, which may loosen the cord domain and affect the stability of the α-helix domain of XynR8. In this research, both substitutions of Gly and Asn with Asp-137 resulted in a substantial increase in the thermostability of enzymes (Fig. 3e, f; Table 3). As the simplest amino acid, Gly plays an important role in protein structures owing to its lack of a β-carbon atom, which permits a substantially greater degree of conformational flexibility than any other residue. Gruber et al. verified the important role of Gly in the loop for the stability of GH11 xylanase from Thermomyces lanuginosus [5]. In this instance, the attractive interaction might disappear when Asp-137 is replaced by Gly-137, causing the cord domain to become quite rigid and resistant to denaturation at high temperatures. After Asn-137 substituted Asp-137 in the mutant, the attractive interaction might be replaced by a repulsive interaction between the Asn-137 and Gln-100, which caused the cord domain to become much more rigid than those of XynR8_CS and the mutant D137G, and improved the enzyme’s stability. The cord domain requires flexibility to allow the accessibility of the xylan substrates; however, the flexibility is also one of the major reasons for the denaturation of the enzyme at high temperature. Consequently, we propose that the stability of the cord domain is important in maintaining enzyme configuration.
The residue Gly-151 was a surface amino acid of XynR8, which was located in the loop between β-strands B10 and B11 and close to the α-helix. As shown in Fig. 3g, a positive-charged amino acid Arg-184 was found to be close to Gly-151. Arg is frequently involved in the formation of a salt bridge, where it is paired with a negatively charged Asp or Glu to create stabilizing hydrogen bonds that could be important for protein stability [2, 4]. In this study, the thermostability of mutants was increased after the Gly-151 was replaced by the Asp-151 or Glu-151. We proposed that a single salt bridge was introduced in the mutants of G151D or G151E which was located between the Asp-151/Glu-151 and the Arg-184, and then stabilized the α-helix domain of XynR8 (Fig. 3h, i). It is easier to form a salt bridge for Glu-151 than Asp-151 in this mutant, because the distance between the amidogen of the Arg-184 and the carboxyl of Glu-151 is shorter than that of Asp-151, because the side chain of Glu contains one more methyl than Asp. Hence, the mutant G151E is more stable than G151D.
Despite enormous efforts, the mechanism underlying the acquirement of xylanase thermostability is still not clear. In this study, by combining random point mutagenesis and site-directed mutagenesis, we obtained a thermostable mutant of xylanase XynR8. The mutant contained three substitutions, which are located at the N-terminus, the cord domain and the α-helix domain of XynR8, respectively, and three important conclusions were proposed. Firstly, the N-terminus (β-strands B1–B6a) of GH11 xylanase is frangible; improvement of its packing density, such as via the substitution of Ile with Val in this domain, may increase the enzyme’s thermostability. Secondly, the stability of the cord domain is important to maintain the GH11 xylanase configuration, and it is also suggested that a reduction in the attractive interaction between the cord domain and other domains may contribute to decrease the enzyme’s entropy and free energy, and to increase its stability. Thirdly, the α-helix domain of GH11 xylanase is also frangible, and introduction of salt bridges in it will increase the enzyme’s stability greatly.
It is well known that a single amino acid substitution could have a remarkable effect on enzyme thermostability, so it would be difficult to compare the sequence of mutation sites of different xylanases from previous studies, and it might also be impossible to reveal credible regularity to guide site-directed mutations of GH11 xylanase on the basis of this comparison’s results. However, though the sequences are different, their 3D structure differences are minor. Consequently, in this study, the location of mutation sites in the 3D structure of different GH11 xylanases from previous studies were compared with each another, and we found that most of the mutations occurred at the N-terminus and α-helix domain. In addition to the two aforementioned domains, the cord domain was also proposed to be responsible for GH11 xylanase’s stability. A chain is no stronger than its weakest link, and improvements in the stability of these three frangible domains may have beneficial effects that increase the thermostability of GH11 xylanase.
References
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22(2):195–201. doi:10.1093/bioinformatics/bti770
Capistran-Licea VM, Millan-Pacheco C, Pastor N (2009) Thermal adaptation strategies used by TBP. Biophys J 96:331
Collins T, Gerday C, Feller G (2005) Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev 29(1):3–23
Georis J, Esteves FD, Lamotte-Brasseur J, Bougnet V, Devreese B, Giannotta F, Granier B, Frere JM (2000) An additional aromatic interaction improves the thermostability and thermophilicity of a mesophilic family 11 xylanase: structural basis and molecular study. Protein Sci 9(3):466–475
Gruber K, Klintschar G, Hayn M, Schlacher A, Steiner W, Kratky C (1998) Thermophilic xylanase from Thermomyces lanuginosus: high-resolution X-ray structure and modeling studies. Biochemistry 37(39):13475–13485. doi:10.1021/bi980864l
Hakulinen N, Turunen O, Janis J, Leisola M, Rouvinen J (2003) Three-dimensional structures of thermophilic beta-1,4-xylanases from Chaetomium thermophilum and Nonomuraea flexuosa: comparison of twelve xylanases in relation to their thermal stability. Eur J Biochem 270(7):1399–1412. doi:10.1046/j.1432-1033.2003.03496.x
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Hokanson CA, Cappuccilli G, Odineca T, Bozic M, Behnke CA, Mendez M, Coleman WJ, Crea R (2011) Engineering highly thermostable xylanase variants using an enhanced combinatorial library method. Protein Eng Des Sel 24(8):597–605. doi:10.1093/protein/gzr028
Kim DY, Han MK, Oh HW, Bae KS, Jeong TS, Kim SU, Shin DH, Kim IH, Rhee YH, Son KH, Park HY (2010) Novel intracellular GH10 xylanase from Cohnella laeviribosi HY-21: biocatalytic properties and alterations of substrate specificities by site-directed mutagenesis of Trp residues. Bioresour Technol 101(22):8814–8821. doi:10.1016/j.biortech.2010.06.023
Kulkarni N, Shendye A, Rao M (1999) Molecular and biotechnological aspects of xylanases. FEMS Microbiol Rev 23(4):411–456
Kumar PR, Eswaramoorthy S, Vithayathil PJ, Viswamitra MA (2000) The tertiary structure at 1.59 A resolution and the proposed amino acid sequence of a family-11 xylanase from the thermophilic fungus Paecilomyces varioti bainier. J Mol Biol 295(3):581–593
Letunic I, Doerks T, Bork P (2011) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Research 40(D1):D302–D305
Liu JR, Yu B, Lin SH, Cheng KJ, Chen YC (2005) Direct cloning of a xylanase gene from the mixed genomic DNA of rumen fungi and its expression in intestinal Lactobacillus reuteri. FEMS Microbiol Lett 251(2):233–241
Liu Y, Kuhlman B (2006) RosettaDesign server for protein design. Nucleic Acids Res 34(suppl 2):W235
Marchler-Bauer A, Lu SN, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke ZX, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang DC, Zhang NG, Zheng CJ, Bryant SH (2011) CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39:D225–D229. doi:10.1093/nar/gkq1189
McHunu NP, Singh S, Permaul K (2009) Expression of an alkalo-tolerant fungal xylanase enhanced by directed evolution in Pichia pastoris and Escherichia coli. J Biotechnol 141(1–2):26–30. doi:10.1016/j.jbiotec.2009.02.021
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428
Miyazaki K, Takenouchi M, Kondo H, Noro N, Suzuki M, Tsuda S (2006) Thermal stabilization of Bacillus subtilis family-11 xylanase by directed evolution. J Biol Chem 281(15):10236–10242. doi:10.1074/jbc.M511948200
Purmonen M, Valjakka J, Takkinen K, Laitinen T, Rouvinen J (2007) Molecular dynamics studies on the thermostability of family 11 xylanases. Protein Eng Des Sel 20(11):551–559. doi:10.1093/protein/gzm056
Sapag A, Wouters J, Lambert C, de Ioannes P, Eyzaguirre J, Depiereux E (2002) The endoxylanases from family 11: computer analysis of protein sequences reveals important structural and phylogenetic relationships. J Biotechnol 95(2):109–131. doi:10.1016/s0168-1656(02)00002-0
Sun JY, Liu MQ, Xu YL, Xu ZR, Pan L, Gao H (2005) Improvement of the thermostability and catalytic activity of a mesophilic family 11 xylanase by N-terminus replacement. Protein Expr Purif 42(1):122–130
Torronen A, Kubicek CP, Henrissat B (1993) Amino acid sequence similarities between low-molecular-weight endo-1,4-beta-xylanases and family H cellulases revealed by clustering analysis. FEBS Lett 321(2–3):135–139. doi:10.1016/0014-5793(93)80094-b
Torronen A, Rouvinen J (1997) Structural and functional properties of low molecular weight endo-1,4-beta-xylanases. J Biotechnol 57(1–3):137–149. doi:10.1016/s0168-1656(97)00095-3
Turunen O, Vuorio M, Fenel F, Leisola M (2002) Engineering of multiple arginines into the Ser/Thr surface of Trichoderma reesei endo-1,4-beta-xylanase II increases the thermotolerance and shifts the pH optimum towards alkaline pH. Protein Eng 15(2):141–145. doi:10.1093/protein/15.2.141
Vardakou M, Dumon C, Murray JW, Christakopoulos P, Weiner DP, Juge N, Lewis RJ, Gilbert HJ, Flint JE (2008) Understanding the structural basis for substrate and inhibitor recognition in eukaryotic GH11 xylanases. J Mol Biol 375(5):1293–1305
Vieille C, Zeikus GJ (2001) Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev 65(1):1
Wang Q, Xia T (2008) Enhancement of the activity and alkaline pH stability of Thermobifida fusca xylanase A by directed evolution. Biotechnol Lett 30(5):937–944. doi:10.1007/s10529-007-9508-1
Winterhalter C, Heinrich P, Candussio A, Wich G, Liebl W (1995) Identification of a novel cellulose-binding domain within the multidomain 120-kDa xylanase xyna of the hyperthermophilic bacterium thermotoga-maritima. Mol Microbiol 15(3):431–444. doi:10.1111/j.1365-2958.1995.tb02257.x
Yadav S, Ahmad F (2000) A new method for the determination of stability parameters of proteins from their heat-induced denaturation curves. Anal Biochem 283(2):207–213
Yang GY, Bai AX, Gao L, Zhang ZM, Zheng BS, Feng Y (2009) Glu88 in the non-catalytic domain of acylpeptide hydrolase plays dual roles: charge neutralization for enzymatic activity and formation of salt bridge for thermodynamic stability. BBA Proteins Proteom 1794(1):94–102. doi:10.1016/j.bbapap.2008.09.007
Yang H, Yao B, Meng K, Wang Y, Bai Y, Wu N (2007) Introduction of a disulfide bridge enhances the thermostability of a Streptomyces olivaceoviridis xylanase mutant. J Ind Microbiol Biotechnol 34(3):213–218
Yang HM, Meng K, Luo HY, Wang YR, Yuan TZ, Bai YG, Yao B, Fan YL (2006) Improvement of the thermostability of xylanase by N-terminus replacement. Chin J Biotechnol 22(1):26–32
You C, Huang Q, Xue HP, Xu Y, Lu H (2010) Potential hydrophobic interaction between two cysteines in interior hydrophobic region improves thermostability of a family 11 xylanase from Neocallimastix Patriciarum. Biotechnol Bioeng 105(5):861–870. doi:10.1002/bit.22623
Zhang S, Zhang K, Chen XZ, Chu X, Sun F, Dong ZY (2010) Five mutations in N-terminus confer thermostability on mesophilic xylanase. Biochem Biophys Res Commun 395(2):200–206. doi:10.1016/j.bbrc.2010.03.159
Zhang ZG, Yi ZL, Pei XQ, Wu ZL (2010) Improving the thermostability of Geobacillus stearothermophilus xylanase XT6 by directed evolution and site-directed mutagenesis. Bioresour Technol 101(23):9272–9278. doi:10.1016/j.biortech.2010.07.060
Acknowledgments
This work was supported by the High-tech Research and Development Program of China (2007AA021302 and 2008AA02Z311).
Author information
Authors and Affiliations
Corresponding author
Additional information
Huping Xue and Jungang Zhou contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Xue, H., Zhou, J., You, C. et al. Amino acid substitutions in the N-terminus, cord and α-helix domains improved the thermostability of a family 11 xylanase XynR8. J Ind Microbiol Biotechnol 39, 1279–1288 (2012). https://doi.org/10.1007/s10295-012-1140-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-012-1140-y