
Abstract
Directed evolution has been used to enhance the catalytic activity and alkaline pH stability of Thermobifida fusca xylanase A, which is one of the most thermostable xylanases. Under triple screened traits of activity, alkaline pH stability and thermostability, through two rounds of random mutagenesis using DNA shuffling, a mutant 2TfxA98 with approximately 12-fold increased k cat/K m and 4.5-fold decreased K m compared with its parent was obtained. Moreover, the alkaline pH stability of 2TfxA98 is increased significantly, with a thermostability slightly lower than that of its parent. Five amino acid substitutions (T21A, G25P, V87P, I91T, and G217L), three of them are near the catalytic active site, were identified by sequencing the genes encoding this evolved enzyme. The activity and stabilizing effects of each amino acid mutation in the evolved enzyme were evaluated by site-directed mutagenesis. This study shows a useful approach to improve the catalytic activity and alkaline pH stability of T. fusca xylanase A toward the hydrolysis of xylan.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Xylanases (EC 3.2.1.8) catalyze the hydrolysis of xylan, a major constituent of hemicellulose. On the basis of primary structure homology, the majority of xylanases have been classified into glycoside hydrolase families 10 and 11 (GH10 and GH11, respectively) (Henrissat and Bairoch 1993). The enzyme has potential economical and environment-friendly applications, including paper and pulp industries and food and feed industries (Bajpai 2004; Polizeli et al. 2005). Xylanases can be used in bleaching of pulp to reduce the use of toxic chlorine-containing chemicals. In addition, it is desirable that xylanases used for biobleaching are stable and active under alkaline conditions at high temperatures. Therefore, attention is focused on the discovery of new xylanases or improving of existing ones in order to achieve stability and activity at high temperature and extreme pH.
Thermobifida fusca (previously Thermomonospora fusca) xylanase A (TfxA) is one of the most thermostable xylanases, retaining 96% of its activity even after 18 h of heating at 75°C (Irwin et al. 1994; Zhang et al. 1998). TfxA consists of a substrate-binding domain and a catalytic domain connected by a linker region (Irwin et al. 1994). The amino acid sequence of the catalytic domain of the enzyme shows that it belongs to family-11 xylanase. However, the practical applications of TfxA to pulp-and-paper industry have been limited, mostly due to its relatively low catalytic activity and stability under alkaline conditions.
Directed evolution of enzyme in vitro is a useful strategy of protein engineering to explore enzyme functions by mimicking key processes of natural evolution. A significant advantage of this method is that neither structural information nor catalytic mechanisms are required to guide the evolution of enzymes. Over the years, the directed evolution method has been proven to be a much more powerful protein engineering tool than conventional rational or random chemical mutagenic approaches and has been frequently used to develop enzymes for developing thermostability and substrate specificity (Gonzalez-Blasco et al. 2000; Meyer et al. 2002; Moore and Arnold 1996; Stemmer 1994). Because the catalytic mechanism and molecular basis of TfxA stability is poorly understood, it is difficult to enhance its properties by rational design. Therefore, to obtain mutants with enhanced enzyme activity and stability, we generated a TfxA catalytic domain (TfxA-cat) by the approach of directed evolution in vitro, and investigated the factors that were associated with the activity, and thermal and alkaline pH stability of this enzyme.
Materials and methods
Cloning of TfxA-cat gene
The DNA encoding of the TfxA-cat was amplified from the plasmid pD731, which was kindly provided by Professor David B. Wilson (Section of Biochemistry, Molecular and Cell Biology, Cornell University, Ithaca, NY, U.S.A.). The PCR primers used were: TfA-N (5′-GTGGGATCCGCCGTGACCTCCAACGAGACCGGGTACC-3′) and TfA-C (5′-GTGCTGCAG TTAGCTGGTGCCCAACGTCACGTTCCAGGACCCGCTGCTCTG-3′). TfA-N includes the BamHI recognition site (underlined), which occurs just before the first codon of the mature enzyme. TfA-C includes the C-terminal region of the catalytic domain; the termination codon is shown in italics and the PstI recognition site is underlined. Amplification of DNA was performed using a MiniCycler (MJ Research, Watertown, MA, U.S.A.) employing the following temperature program: 5 min at 98°C, 25 cycles of 1 min at 98°C, 1 min at 65°C, 1 min at 72°C, followed by 10 min at 72°C. After the first 5 min denaturation step the temperature program was paused, TaKaRa LA Taq (1.25 units; Takara Shuzo, Shiga, Japan) was added to the reaction mixture (25 μl), and heating was immediately continued. Using this method, GC-rich target genes were efficiently amplified. The PCR products were subcloned into pCR2.1 (Invitrogen, Carlsbad, CA) and the nucleotide sequences were confirmed. The TfxA-cat gene was cleaved by BamHI and PstI and ligated with pQE30 (Qiagen, Hilden, Germany) using the BamHI and PstI sites to obtain the TfxA-cat plasmid vector for further expression and DNA shuffling.
DNA shuffling of the TfxA-cat gene
Ten micrograms TfxA-cat plasmid DNA were re-suspended in 100 μl sterile TE buffer (10 mM Tris/HCl, 1 mM EDTA, pH 8.0). The mixture was then treated with DNase I (Takara) for fragmentation in Tris/HCl buffer (pH 7.4) containing 10 mM MnCl2 and 25 mM MgSO4 (25°C, 10 min). After inactivating DNase I by heating at 90°C for 10 min, the digests were electrophoresed on a 2% (w/v) agarose gel. DNA fragments (<100 bp) were extracted from the gel and recovered in 50 μl of distilled water. The purified DNA fragments were then subjected to 40 cycles of the first PCR reaction without any oligonucleotide primers as described above, except that annealing was performed at 45°C.
PCR was carried out using 2.5 μl of the fragment reassembly products using the primers 30BU (5′-CTATGAGAGGATCGCATCACCATCACCATCACGGATCC-3′) and 30PL (5′-CAGGAGTCCAAGCTCAGCTAATTAAGCTTGGCTGCAG-3′), which correspond to the outer region of the BamHI and PstI sites (underlined) of pQE30 respectively. The PCR products were digested with BamHI and PstI, purified by agarose-gel electrophoresis, then ligated into pQE30 between the BamHI and PstI sites. The recombinant plasmids were transformed into E. coli M15 (Qiagen, Hilden, Germany) and plated on to Luria–Bertani (LB) agar containing 50 μg ampicillin/ml, 50 μg kanamycin/ml and 0.5% 4-O-methyl-d-glucurono-d-xylan–Remazol Brilliant Blue R (RBB-xylan; Sigma). Colonies capable of acting on RBB-xylan were selected for screening.
Screening of the mutant libraries
TfxA xylanase variants with improved alkaline pH stability under high temperature were primarily screened by using a pH 9.0 RBB-xylan agar plate. Agar plates contained 0.5% RBB-xylan and 1.5% agar in 50 mM Tris/HCl, pH 9.0. Variants from a mutant library were tooth-picked to an alkaline RBB-xylan agar plate with control colonies. In the first round, the RBB-xylan plate was incubated at 60°C for 1 h, then overnight at room temperature. ‘Successful’ colonies were identified by their ability to form a distinct halo in the RBB-xylan-containing agar. Wild-type TfxA completely lost activity after the above-mentioned steps and no distinct halo occurred on the agar plates. In the second round, a mutant library was incubated under conditions where the best mutant selected in the first round lost activity completely: pH 9.0 RBB-xylan agar plate, 60°C for 2 h.
Site-directed mutageneses
Site-directed mutageneses were performed, using the ExSite PCR-based site-directed mutagenesis kit (Stratagene Cloning Systems, Inc., La Jolla, Calif.). Complementary primers for PCR containing the desired DNA changes were constructed and included in reaction mixtures in which 40 ml distilled H2O, 5 ml 10× pfu-turbo polymerase buffer, 1 ml forward primer (125 ng/ml), 1 ml reverse primer (125 ng/ml), 1 ml dNTPs (10 mM), 1 ml plasmid (10–50 ng), and 1 ml pfu-turbo polymerase (2.5 U/ml) were combined in a total reaction volume of 50 ml. Thermocycler settings used were the same as described above, except DpnI (1 ml) was added to the reaction. The resulting plasmids from the final reaction mixture were transformed into competent cells of E. coli JM109, and the resultant colonies were DNA sequenced to identify the clones containing the desired substitutions.
Enzyme production and purification
Recombinant E. coli was grown at 37°C in 100 ml of LB medium containing ampicillin and kanamycin. Protein production was induced by adding 0.4 mM IPTG to the culture media when the OD660 reached 0.6. Cultivation was continued overnight. Cells were harvested by centrifugation (6,000 × g, 10 min at 4°C). About 2.5 g cells were subjected to ultrasonication for enzyme extraction. The cell-free lysates were then centrifuged (10,000 × g, 10 min at 4°C) and each lysate was charged with 1 ml Ni-NTA resin, which bound the target proteins. The resin was then packed into 1 ml column and eluted with a linear gradient of 250 mM imidazole in 50 mM Sodium phosphate buffer, pH 8.0 at 0.8 ml/min using an FPLC system. The active fractions were combined and dialyzed against 1 l 20 mM CHES buffer, pH 9.0 or 20 mM MES buffer pH 6.0 overnight at 4°C. The dialyzed enzyme solutions were reloaded onto a Q-Sepharose or a SP-Sepharose column (8 ml) previously equilibrated with either 20 mM CHES buffer, pH 9.0 or 20 mM MES buffer pH 6.0 and the enzymes were eluted with a linear gradient of 0.5 M or 1.0 M NaCl at 0.5 ml/min. The homogeneity of the purified enzyme fractions was monitored by SDS-PAGE, and the relevant fractions were pooled and dialyzed against de-ionized water.
Enzyme assay
Crude and purified enzymes were assayed quantitatively using 0.2% RBB-xylan (Sigma, Germany) as a substrate. Concentrated enzyme solution was diluted with assay buffer, 50 mM McIlvaine buffer, pH 7.0 (0.1 M citric acid and 0.2 M Na2HPO4) (Kaneko et al. 2004), to give 0.01 μg protein/μl. 5 μl of diluted enzyme was applied to a pre-warmed mixture of 20 μl of 50 mM McIlvaine buffer pH 7.0 and 25 μl of 0.4% aqueous solution of RBB-xylan and incubated at 50°C for 15 min. The reaction was stopped by adding of 100 μl ethanol and incubating at room temperature at least for 15 min. The precipitated residual substrate was collected by centrifugation at 12,000 × g for 2 min and the OD of the supernatant was measured at 595 nm against respective substrate blanks.
pH and thermal stability
pH stability was determined by exposing purified enzyme (10 ± 2 μg of protein/ml) to 50 mM Tris/HCl buffer, pH 9.0 at 50°C for various periods of time. The residual activities of the treated enzymes were determined. Thermostability was tested by heating enzyme samples (10 ± 2 μg of protein/ml) at 70°C in 50 mM McIlvaine buffer, pH 7.0 for various periods of time. The residual activity of the treated enzymes was then measured.
Analysis of kinetic parameters
Steady-state kinetic parameters were determined as described by Lawson et al. (1996). Soluble birchwood xylan, prepared as described previously (Dupont et al. 1998), was used as a substrate. K m and k cat values were determined using substrate concentrations ranging from 0.4 to 10 mg xylan/ml in 30% of 0.2 M McIlvaine buffer, pH 7.0. After incubation at 50°C for 30 min, the production of reducing sugar was measured by using p-hydroxybenzoic acid hydrazide (Lever 1972). Protein concentration was measured using DC Protein Assay Reagents (Bio-Rad) with BSA as the standard.
Results
Screening for enhanced stability and activity of TfxA in alkaline pH solution
The recombinant enzyme TfxA-cat consists of 189 amino acid residues and exhibits the same activity as native enzyme when soluble xylan is used as the substrate. TfxA is composed of a catalytic domain, a binding domain and a linker region (Irwin et al. 1994). Removal of the substrate-binding domain from the native enzyme has no effect on the hydrolysis of soluble xylan (Irwin et al. 1994). TfxA-cat was functionally expressed in E. coli and showed similar properties to those reported for the native enzyme (Irwin et al. 1994).
To obtain evolved TfxA with increased stability and without activity loss under alkaline conditions at high temperatures, a two-step evolutionary engineering approach was applied to TfxA. In the primary round of mutagenesis and screening, a mutant library of TfxA was prepared with DNA shuffling and screened on alkaline RBB-xylan plates (pH 9.0) and treated as described above. Theoretically, colonies capable of acting on RBB-xylan after this treatment possessed thermal and alkaline pH stability, because these incubation conditions inactivated TfxA. In cases where the recombinant enzyme is not stable at 60°C or pH 9.0, only a small halo is produced. A small halo may also indicate low specific activity, even if the enzyme is relatively stable at 60°C and pH 9.0. Using this screening method, the variants with improved thermal stability in an alkaline pH range compared to the parental enzyme were easily screened.
After the first round of random mutagenesis using DNA shuffling, the parental TfxA enzyme did not form a detectable clear zone on the RBB-xylan plate. Variants with increased halo formation were presumed to have higher alkaline pH and thermal stability than the parental enzyme. Therefore three positive clones containing TfxA variants (1TfxA6, 1Tfx302, and 1Tfx890) resistant to pH 9.0 alkaline and 60°C high temperature conditions were isolated from approximately 1,000 colonies. Plasmids encoding each mutant TfxA were isolated from three clones and digested with BamHI and PstI. The digested DNA fragments were pooled and recombined in vitro to accumulate beneficial mutations. The resultant products of DNA shuffling were subcloned in order to prepare a mutant library for the second evolutionary cycle. After the second mutant library was screened on pH 9.0 RBB-xylan plate and incubated at 60°C for 2 h. The most alkaline and thermostable variant (2TfxA89) was identified from screening of approximately 1,010 clones. All variants from the screening were sequenced to identify the locations of the substituted amino acid residues.
Sequence analysis of evolved TfxA variants
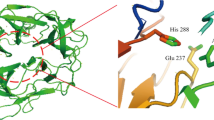
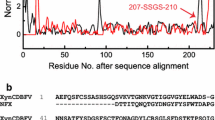
DNA sequence analysis revealed that 1TfxA6, 1TfxA302, and 1TfxA890, obtained in the first round of shuffling, contained single or double amino acid substitutions (V87P and G217L for 1TfxA6, V87P for 1TfxA302, T21A and V87P for 1TfxA890) (Fig. 1). These three clones were used as parental clones in the second round of shuffling. More than 1,000 clones from the second-generation library were screened on pH 9.0 RBB-xylan plate after heat treatment at 60°C for 2 h. Clone 2TfxA98 exhibited the highest stability. The second round of DNA shuffling introduced the additional two mutations G25A and J91T into 2TfxA98, along with 1TfxA6, 1TfxA302, and 1TfxA890 mutations (Fig. 1). The mutants V87P, I91T, and G217L are located in the vicinity of the catalytic triad (Glu88 and Glu216) of TfxA (Irwin et al. 1994). Interestingly, all mutant TfxA genes selected for their high activity toward alkaline pH RBB-xylan contained the same V87P substitution. This indicates that V87P substitution played an important role in the stability and activity of TfxA and this substitution may be responsible for converting a moderately active TfxA parent protein into the highly active chimeras. Apparently, the greater activity of chimeras depends not on the single site substitution, but on the directed evolution of this change in concert with the appropriate context provided by the rest of the protein.

Lineage of TfxA variants. Amino acid substitution accumulated by two generations of mutants are shown. * denotes same amino acid substitution in all mutants
Alkaline pH stability and thermostability of the evolved 2TfxA98 mutant
The recombinant TfxA and the mutant 2TfxA98 xylanases were purified to homogeneity detected by SDS-PAGE (data not shown) using a nickel-chelating FPLC column. The residual enzyme activity was traced in the presence of 50 mM Tris/HCl buffer, pH 9.0 at 50°C for various periods of time. The mutant 2TfxA98 xylanase showed about 80% of the initial activity after 180 min of incubation, while the recombinant TfxA almost completely lost its activity under the same conditions (Fig. 2A). The residual activities of both enzymes after heating at 70°C are shown in Fig. 2B. Both recombinant TfxA and mutant 2TfxA98 were stable at 70°C during 30 min incubation, although the mutant was slightly less stable than the parent at 70°C after 30 min of incubation. Therefore, the mutant 2TfxA98 inherits its thermostability from parent TfxA and enhances its alkaline pH stability through DNA shuffling.

Alkaline pH and thermal stability curves of purified parent TfxA and mutant 2TfxA98. The parent and mutant enzymes were incubated at 50°C in 50 mM Tris/HCl buffer, pH 9.0 (A) or heated at 70°C in 50 mM McIlvaine buffer, pH 7.0 (B). The residual activity was measured at 50°C with 5 mg/ml soluble xylan in 50 mM McIlvaine buffer, pH 7.0. Residual activity is expressed as a percentage of the maximum of enzyme activity (0.26 U/ml) under standard assay conditions. All data plotted are averages for three independent experiments
Activities of the evolved 2TfxA98 mutant
Kinetic parameters of the recombinant TfxA and mutant 2TfxA98 xylanases are shown in Table 1. The K m value of 2TfxA98 was decreased 4.5-fold compared to the wild type. The catalytic constant (k cat) was 2.6-fold higher than that of TfxA. Therefore the k cat/K m ratio of mutant 2TfxA98 was increased 12-fold compared to the parental enzyme. This indicates that the mutant 2Tfx98 had significantly improved catalytic activities in performing the xylan hydrolysis.
Reducing-sugar productivity from soluble xylan
The xylan-hydrolyzing activities of the parent and mutant enzymes were determined at various temperatures and pH 9.0 buffer (Fig. 3). The mutant 2TfxA98 xylanase was most active at 75°C, almost the same as the parental TfxA. However, 2TfxA98 produced 1.9 times more reducing sugar than TfxA at 75°C of pH 9.0, indicating that the catalytic activity of 2TfxA98 was significantly improved under conditions of alkaline and high temperature. The evolved enzyme 2TfxA98 can meet the requirements of biobleaching by its stability and activation under conditions of alkaline and high temperatures.

Reducing-sugar productivity of purified parent TfxA and mutant 2TfxA98. Enzyme reactions were performed in 50 mM McIlvaine buffer, pH 7.0. The data plotted are averages for three independent experiments
Amino acid substitutions in the mutant 2TfxA98 xylanase
Mutated amino acid residues in 2TfxA98 were identified by sequencing the corresponding gene. 2TfxA98 was found to possess five mutations, T21A, G25P, V87P, J91T, and G217L. To evaluate the contribution of each amino acid change in increasing catalytic activity and alkaline pH stability, site-directed mutagenesis was conducted to incorporate each mutation into the parental TfxA, and the stability of the resulting mutants were tested. V87P significantly improved both the activity and alkaline pH stability of 2TfxA98 (Table 2). G217L also increased activity and alkaline pH stability to some extent. The mutations, T21A and G25P, mainly led to increase in alkaline pH stability without improving the specific activity. The other mutation I91T resulted in the main contribution to catalytic activity rather than alkaline pH stability in 2TfxA98.
Discussion
The present study demonstrated that DNA shuffling is applicable for direct evolution of thermostable xylanase with improved catalytic activity. During two subsequent rounds of DNA shuffling, mutant 2TfxA98 were selected for thermal and alkaline pH stability and also improved catalytic properties for the hydrolysis of RBB-xylan.
To date, the crystal structures of family 11 xylanases are available from several organisms: Trichoderma harzianum (Campbell et al. 1993), Bacillus circulans (Sidhu et al. 1999; Wakarchuk et al. 1994), Trichoderma reesei (Törrönen et al. 1994; Törrönen and Rouvinen 1995), Aspergillus niger (Krengel and Dijkstra 1996), Thermomyces lanuginosus (Gruber et al. 1998), Aspergillus kawachii (Fushinobu et al. 1998), Bacillus agaradhaerens (Sabini et al. 1999), Paecilomyces varioti (Kumar et al. 2000), and Dictyoglomus thermophilum (McCarthy et al. 2000). Three of these, T. lanuginosus, P. varioti, and D. thermophilum are from thermophilic organisms. A disulphide bridge has been suggested as one reason for the enhanced thermal stability of T. lanuginosus and P. varioti xylanases (Gruber et al. 1998; Kumar et al. 2000). A greater proportion of polar surface and a slightly extended C-terminus together with an extension of β-strand A5 are thought to increase the stability of D. thermophilum xylanase (McCarthy et al. 2000; Morris et al. 1998). However, the structural basis for the thermostability of family 11 xylanases is not well understood.
Previous studies revealed several minor modifications responsible for the increased thermal stability of family 11 xylanases: (a) a higher Thr:Ser ratio (b) an increased number of charged residues, especially Arg, resulting in enhanced polar interactions, and (c) an improved stabilization of secondary structures due to an increased number of residues in the β-strands and stabilization of the α-helix region. Some members of family 11 xylanases have a unique feature to improve their stability, such as a higher number of ion pairs or aromatic residues on protein surface, a more compact structure, tighter packing, and insertions at some regions resulting in enhanced interactions. These changes increase protein rigidity, a property associated with enhanced stability. Several studies have demonstrated that the highly thermophilic xylanases have a great number of side chain-side chain polar interactions and several salt bridges. There could be a trend in xylanase structures that acidophilic xylanases have few salt bridges than alkalophilic xylanases. It is possible that alkaline and thermal adaptation use similar mechanisms for improving stability.
Thermobifida fusca xylanase A is one of the most thermostable xylanases (Irwin et al. 1994). In this study, we successfully improve the catalytic activity and alkaline pH stability of TfxA by DNA shuffling. Structural and sequence comparisons of the 2TfxA98 mutant with the TfxA parent revealed five amino acid substitutions. Three substitutions, V87P, I91T, and G217L are located in the vicinity of the catalytic triad (Glu88 and Glu216) of TfxA (Irwin et al. 1994). Interestingly, all mutant TfxA genes with high activity toward alkaline pH RBB-xylan contained the same V87P substitution. This indicates that V87P substitution played an important role in stability and activity of TfxA and this substitute may be responsible for converting a moderately active TfxA parent protein into the highly active chimeras. Moreover, V87P mutation resulted in a significantly improved enzyme activity and alkaline pH stability, and G217L also increased activity and alkaline pH stability to some extent. I91T gave rise to increments in enzyme activity rather than alkaline pH stability of 2TfxA98. Another two substitutions (T21A and G25P) mainly increased in alkaline pH stability without improving the specific activity of the enzyme. Further studies based on site-directed mutagenesis and the three-dimensional structure will elucidate the role of each mutation in controlling catalytic activity and alkaline pH stability.
In summary, DNA shuffling was used to improve the catalytic activity and alkaline pH stability of T. fusca xylanase A toward the hydrolysis of xylan. An efficient method was used to screen a library of about 2,000 clones leading to the identification of four highly selective mutants with 5–12-fold enhanced specific activity toward the substrate of interest. The alkaline pH stability of the selected mutant was also significantly improved compared with that of parental enzyme. Further study of the mutant enzyme should expand our understanding of the structure-function relationship for the valuable biocatalyst.
References
Bajpai P (2004) Biological bleaching of chemical pulps. Crit Rev Biotechnol 24:1–58
Campbell RL, Rose DR, Wakarchuk WW, To R, Sung W, Yaguchi M (1993) Trichoderma reesei cellulases and other hydrolases. In: Suominen P, Reinikainen T (eds) Foundation for biochemical and industrial fermentation research. Espoo, Finland, pp 63–72
Dupont C, Roberge M, Shareck F, Morosoli R, Kluepfel D (1998) Substrate-binding domains of glycanases from Streptomyces lividans: characterization of a new family of xylan-binding domains. Biochem J 15:41–45
Fushinobu S, Ito K, Konno M, Wakagi T, Matsuzawa H (1998) Crystallographic and mutational analyses of an extremely acidophilic and acid-stable xylanase: biased distribution of acidic residues and importance of Asp37 for catalysis at low pH. Protein Eng 11:1121–1128
Gonzalez-Blasco G, Sanz-Aparicio J, Gonzalez B, Hermoso JA, Polaina J (2000) Directed evolution of beta-glucosidase A from Paenibacillus polymyxa to thermal resistance. J Biol Chem 275:13708–13712
Graddis TJ, Remmele RL Jr, McGrew JT (2002) Designing proteins that work using recombinant technologies. Curr Pharm Biotechnol 3:285–297
Gruber K, Klintschar G, Hayn M, Schlacher A, Steiner W, Kratky C (1998) Thermophilic xylanase from Thermomyces lanuginosus: High-resolution X-ray structure and modeling studies. Biochemistry 37:13475–13485
Henrissat B, Bairoch A (1993) New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 293:781–788
Irwin D, Jung ED, Wilson DB (1994) Characterization and sequence of a Thermomonospora fusca xylanase. Appl Environ Microbiol 60:763–770
Kaneko S, Ichinose H, Fujimoto Z, Kuno A, Yura K, Go M, Mizuno H, Kusakabe L, Kobayashi H (2004) Structure and function of a family 10 beta-xylanase chimera of Streptomyces olivaceoviridis E-86 FXYN and Cellulomonas fimi Cex. J Biol Chem 279:26619–26626
Krengel U, Dijkstra BW (1996) Three-dimensional structure of endo-1,4-β-xylanase I from Aspergillus niger: Molecular basis for its low pH optimum. J Mol Biol 263:70–78
Kumar PR, Eswaramoorthy S, Vithayathil PJ, Viswamitra MA (2000) The tertiary structure at 1.59 Å resolution and the proposed amino acid sequence of a family-11 xylanase from the thermophilic fungus Paecilomyces varioti Bainier. J Mol Biol 295:581–593
Lawson SL, Wakarchuk WW, Withers SG (1996) Effects of both shortening and lengthening the active site nucleophile of Bacillus circulans xylanase on catalytic activity. Biochemistry 35:10110–10118
Lever M (1972) A new reaction for colorimetric determination of carbohydrates. Anal Biochem 47:273–279
McCarthy AA, Morris DD, Bergquist PL, Baker EN (2000) Structure of TRX IB, a highly thermostable β-1,4-xylanase from Dictyoglomus thermophilum Rt46B.1, at 1.8 Å resolution. Acta Crystallogr D56:1367–1375
Meyer A, Schmid A, Held M, Westphal AH, Rothlisberger M, Kohler HP, van Berkel WJ, Witholt B (2002) Changing the substrate reactivity of 2-hydroxybiphenyl 3-monooxygenase from Pseudomonas azelaica HBP1 by directed evolution. J Biol Chem 277:5575–5582
Moore JC, Arnold FH (1996) Directed evolution of a p-nitrobenzyl esterase for aqueous-organic solvents. Nat Biotechnol 14:458–467
Morris DD, Gibbs MD, Chin CW, Koh MH, Wong KKY, Allison RW, Nelson PJ, Bergquist PL (1998) Cloning of the TRX IB gene from Dictyoglomus thermophilum Rt46B.1 and action of the gene product on Kraft pulp. Appl Environ Microbiol 64:1759–1765
Polizeli ML, Rizzattim AC, Monti R, Terenzi HF, Jorge JA, Amorim DS (2005) Xylanases from fungi: properties and industrial applications. Appl Microbiol Biotechnol 67:577–591
Sabini E, Sulzenbacher G, Dauter M, Dauter Z, Jorgensen PL, Schulein M, Dupont C, Davies GJ, Wilson KS (1999) Catalysis and specificity in enzymatic glycoside hydrolysis: a 2,5B conformation for the glycosyl-enzyme intermediate revealed by the structure of the Bacillus agaradhaerens family 11 xylanase. Chem Biol 6:448–455
Sidhu G, Withers SG, Nguyen NT, McIntosh LP, Ziser L, Brayeer GD (1999) Sugar ring distortion in the glycosyl-enzyme intermediate of a family G/11 xylanase. Biochemistry 38:5346–5354
Stemmer WP (1994) Rapid evolution of a protein in vitro by DNA shuffling. Nature 370:389–391
Törrönen A, Harkki A, Rouvinen J (1994) Three-dimensional structure of endo 1,4-β-xylanase II from Trichoderma reesei: two conformation states in the active site. EMBO J 13:2493–2501
Törrönen A, Rouvinen J (1995) Structural comparison of two major endo-1,4-xylanases from Trichoderma reesei. Biochemistry 34:847–856
Wakarchuk WW, Campbell RL, Sung WL, Davoodi J, Yaguchi M (1994) Mutational and crystallographic analyses of the active-site residues of the Bacillus circulans xylanase. Protein Sci 3:467–475
Zhang Z, Wang Y, Ruan J (1998) Reclassification of Thermomonospora and Microtetraspora. Int J Syst Bacteriol 48:411–422
Acknowledgements
We thank Ms. Ying Li for the assistance with the DNA sequence analysis and mutant library construction. This work was financially supported by Shanghai Pujiang Program to Qin Wang.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Q., Xia, T. Enhancement of the activity and alkaline pH stability of Thermobifida fusca xylanase A by directed evolution. Biotechnol Lett 30, 937–944 (2008). https://doi.org/10.1007/s10529-007-9508-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-007-9508-1