
Abstract
Kluyveromyces marxianus is thermotolerant yeast that is able to utilize a wider range of substrates and has greater thermal tolerance than most other yeast species. K. marxianus can assimilate xylose, but its ability to produce ethanol from xylose in oxygen-limited environments is poor. In the present study, the K. marxianus xylose reductase (KmXR) gene (Kmxyl1) was cloned and the recombinant enzyme was characterized to clarify the factors that limit xylose fermentation in K. marxianus NBRC1777. KmXR is a key enzyme in the xylose metabolism of K. marxianus, which was verified by disruption of the Kmxyl1 gene. The Km of the recombinant KmXR for NADPH is 65.67 μM and KmXR activity is 1.295 U/mg, which is lower than those of most reported yeast XRs, and the enzyme has no activity with coenzyme NADH. This result demonstrates that the XR from K. marxianus is highly coenzyme specific; combined with the extremely low XDH activity of K. marxianus with NADP+, the limitation of xylose fermentation is due to a redox imbalance under anaerobic conditions and low KmXR activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
d-Xylose is one of the main hydrolysis products of lignocellulosic biomass and is the second most abundant fermentable material [6, 19, 33, 38]. In contrast to the efficient glucose fermentation in yeast, xylose fermentation is challenging because only a few ethanol-producing microorganisms can readily ferment xylose even though many microorganisms utilize xylose as a carbon source [19].
Highly efficient xylose fermentation could improve the utilization of biomass [16]. To utilize and ferment xylose, the xylose should first be converted into xylulose. Then, xylulose is phosphorylated into xylulose 5-phosphate by xylulokinase (XK), and the xylulose 5-phosphate is metabolized via the pentose phosphate pathway [29, 32]. In bacteria, the d-xylose is converted to d-xylulose directly by xylose isomerase (XI); in eukaryotes such as yeasts and filamentous fungi, d-xylose is converted to d-xylulose through reduction and oxidation, which is catalyzed by xylose reductase (XR) and xylitol dehydrogenase (XDH), respectively [16].
XR is the first enzyme in xylose metabolism and it catalyzes xylose to xylitol in eukaryotes. Generally, poor ethanol production from xylose in yeasts is due to the accumulation of xylitol and the inability to catalyze xylitol to xylulose further during anaerobic fermentation [4]. The main limiting factor is the imbalance of redox, which is caused by the difference in the preferred coenzyme of XR (NADPH) and XDH (NAD+) [37].
K. marxianus can grow at temperatures as high as 52°C [2]. Even at 45°C, K. marxianus still can produce ethanol well [14, 22, 23]. K. marxianus strains are able to utilize a wider range of substrates and exhibit greater thermal tolerance than most other yeast [28]. One of the important potential applications of these strains is the simultaneous saccharification and fermentation (SSF) of cellulosic biomass [5, 8]. The optimal temperature of cellulases used in SSF is 45–50°C; although most yeast cannot survive at such temperatures, K. marxianus strains tolerate higher SSF temperatures. The high fermentation temperature allows more rapid and efficient enzymatic cellulose hydrolysis [3, 34]. Some K. marxianus strains are able to consume xylose and produce ethanol in oxygen-limited environments. K. marxianus IMB4 produces ethanol from xylose with 23% of the theoretical yield at 45°C [2] and 2.08 g/l ethanol at 40°C [34]. Other K. marxianus strains can produce 5.6 g/l ethanol from 20 g/l xylose at 35°C or produce 0.8–1.2 g/l ethanol with 10 g/l xylose [2, 4]. However, these results show that the xylose fermenting ability of K. marxianus is not high [2, 4, 21, 34]. To lay the foundation for the further improvement of xylose fermentation in K. marxianus, the factors that limit fermentation in this yeast need to be determined.
In the present study, we cloned the K. marxianus XR gene (KmXR; Kmxyl1) and characterized the recombinant enzyme of K. marxianus NBRC1777. The function of the Kmxyl1 in the xylose metabolism of K. marxianus was confirmed by disruption of xyl1 in the genome. Combined with the properties of XDH in the yeast, we confirmed that coenzyme bias and low activity were the main reasons that hinder efficient xylose fermentation in K. marxianus.
Materials and methods
Reagents and microorganisms
All chemicals used were of analytical grade or higher. d-Xylose and dNTPs were obtained from Sangon Biotech Co. (Shanghai, China); d-galactose, d-arabinose, d-mannose, Coomassie Brilliant Blue R-250, NADH, NADPH, and agar A were obtained from Bio Basic, Inc. (Toronto, Canada). Restriction enzymes and modifying enzymes were obtained from Fermentas Life Sciences (Fermentas China, Shengzheng, China). The yeast extract and peptone were purchased from Oxoid (Netherlands). K. marxianus NBRC1777 was from NBRC (Tokyo, Japan). K. marxianus strains were cultured in yeast extract/peptone-dextrose (YPD) medium. Escherichia coli XL10-Gold and BL21 star (DE3) were used for cloning and expression, respectively, and were grown in Luria–Bertani (LB) medium.
Cloning of Kmxyl1 gene from K. marxianus NBRC1777
K. marxianus NBRC1777 was cultured in YPD medium at 37°C for 24 h, after which the genomic DNA was extracted, as described before, with glass beads and vortex to break the yeast cell [15]. The part sequence of the XR gene (Kmxyl1) was amplified by PCR with a pair of degenerated primers (XR-F1 and XR-R1 in Table 1), which were designed based on conserved amino acid sequence of XR (EKYPPGFY and RFNDPWD/EW) (Fig. 1).

Nucleotide sequence of the xyl1 gene from K. marxianus and its deduced amino acid sequence. The untranslated and coding regions are represented by small letters and capital letters, respectively, and the asterisk indicates the stop codon. Underlined nucleotides represent the proposed TATA box in the 5′ region and the polyadenylation site in the 3′ region. Arrows indicate the annealing position and direction of primer extension for TAIL-PCR; the open box shows the position on which the degenerated primers design is based
After getting the partial sequence of Kmxyl1, the unknown flanking sequences were amplified by thermal asymmetric interlaced PCR (TAIL-PCR) [20]. The specific primers for the 3′ terminal and 5′ flanking sequence were XR-FS1, XR-FS2, XR-FS3 and XR-RS1, XR-RS2, XR-RS3, respectively. The arbitrary degenerate (AD) primers were AD1, AD2, AD3, and AD4. The arbitrary primer for the second and third round TAIL-PCR was AC1 (Table 1). The PCR cycle conditions were the same as those described by Liu [20]. Amplified flanking sequences were cloned into pMD18-T and validated by sequencing. The full-length sequence of Kmxyl1 was obtained through the assembly all the sequences.
Protein expression and purification
The ORF of Kmxyl1 was amplified by PCR with primers XR-NDE and XR-HIND (Table 1) from the genomic DNA of K. marxianus NBRC1777. The PCR products and plasmid pET-21c were double digested with Nde I and Hind III, and then ligated to construct plasmid pET-21XR. This plasmid was transformed into E. coli BL21 (DE3). One of the positive colonies was inoculated into liquid LB medium that contained 100 μg/ml ampicillin and was cultured at 37°C. When OD600 reached 0.4, isopropyl-α-d-thiogalactopyranoside (IPTG) was added with a final concentration of 0.02 mM and incubation was continued for 4 h. Subsequently, the cells were harvested and resuspended in Tris–HCl buffer (50 mM Tris–HCl and 25 mM sodium chloride at pH 8.0). Furthermore, the cells were lysed by sonication (Vibra-Cell VC505, Connecticut, USA) for 8 min at 40% power. The supernate and lysate pellet fractions were separated by centrifugation at 7,000 × g for 30 min. Afterwards, the recovered supernate was loaded onto a Ni2+-chelating column (Qiagen, USA) and washed according to the manual. Subsequently, the purified proteins were eluted from the column by a gradient of 1 to 250 mM imidazole in 50 mM Tris–HCl, 300 mM sodium chloride buffer (pH 8.0). All samples from the column were analyzed by SDS-PAGE and the purified recombinant KmXR was desalted by dialyzing against 100 mM phosphate buffer (pH 8.0). The purified protein was stored with 20% glycerol at –80°C. Protein concentrations were determined via the Bradford method with bovine serum albumin (BSA) as the protein standard [9].
XR activity assay
The activity of KmXR was determined according to a previously described method [36, 38] using a spectrophotometer to monitor the change in A340 upon oxidation of NAD(P)H. Unless indicated otherwise, the KmXR assay mixture (1.0 ml) for the reaction contained 100 mM phosphate buffer (pH 7.0), 200 μM NAD(P)H, 200 mM xylose, and enzyme solution (0.1 ml). This reaction mixture was allowed to stand for 1 min to eliminate the endogenous oxidation of NADPH, and the reaction was started by the addition of 0.1 ml of substrate. One unit of enzyme activity is defined as the amount of enzyme required to oxidize 1 μmol of NADPH per min under the specified conditions.
Optimal temperature and thermostability
The optimal temperature was studied by measuring KmXR activity at temperatures ranging from 15 to 40°C. To determine the thermal stability of KmXR, the enzyme was incubated at 30–60°C for 10 min. The retained enzyme activity was measured as described in the XR activity assay.
Optimal pH
To determine the optimal pH, enzyme activity was measured at pH levels from 5.0 to 9.0. The following buffers were used to adjust the pH of the reaction mixture: 100 mM acetic acid–sodium acetate buffer (for pH 5.0–5.5), 100 mM phosphate buffer (for pH 6.0–7.5), and 100 mM Tris–HCl buffer (for pH 8.0–9.0).
Substrate specificity
The substrate specificity of KmXR was measured by replacing 200 mM d-xylose with 200 mM d-glucose, d-ribose, d-galactose, or d-arabinose as the substrate in the reaction mixture. The other conditions were as described in the KmXR activity assay.
Effects of metal ion and DTT
The purified recombinant KmXR was dialyzed in 100 mM phosphate buffer (pH 7.0) containing 10 mM EDTA at 4°C overnight to remove the metal ions, after which the enzyme was dialyzed in 100 mM phosphate buffer (pH 7.0, EDTA-free) for 9 h at 4°C to remove EDTA. Thereafter, the activity of the recombinant KmXR was measured in the presence of 1 mM CuSO4, 1 mM MnCl2, 1 mM MgSO4, 1 mM CoCl2, 1 mM NiSO4, 1 mM ZnCl2, 1 mM FeCl3, 1 mM CaCl2, or 1 mM DTT in the reaction mixture [100 mM phosphate buffer (pH 7.0), 0.02 mM NADPH, 200 mM xylose].
Kinetics assay
The kinetic parameters for xylose were obtained from the initial velocity measured at a constant NADPH concentration (0.02 mM) with xylose of various concentrations. The kinetic parameters for NADPH were determined using a constant xylose concentration (0.2 M) and varying NADPH concentrations. All measurements were performed at optimal pH and optimal temperature. All results were performed in triplicate and expressed as mean values; the bars in the figures indicate the ranges of the errors.
Kmxyl1 disruption
Homologous recombination [1, 10] was used to disrupt Kmxyl1 in K. marxianus YHJ010 [15], which is the ura3, trp1, and leu2 auxotroph strain from K. marxianus NBRC1777. The trp1 gene of Saccharomyces cerevisiae was amplified from the plasmid YEGAP [13] with the primers TRP-SAL-F/TRP-SAL-R (Table 1). The amplified trp1 and pET21XR were digested with SalI and ligated together to produce pET21XR-TRP. The Kmxyl1 gene with inserted trp1 was amplified by PCR using the primers XR-F/XR-R (Table 1) and transformed into K. marxianus YHJ010. The positive colonies were screened on synthetic dextrose (SD) medium containing uracil and leucine. The obtained colonies were further selected in the SD medium with d-glucose, d-xylose (SDX1), or d-xylitol (SDX2) as the sole carbon sources.
Site-directed mutagenesis of the Ser-280 in KmXR
From the alignment of xylose reductase amino acid sequence and the mutation results reported by other research, Ser-280 in KmXR which is Asn at this position in most XR reported was estimated to be the amino acid residue leading to the relativity low activity and coenzyme specific of KmXR. Site-directed mutagenesis was used to verify its function. The mutations were constructed with overlap extension PCR with the primers STOD-F/STOD-R (for S280D) and STON-F/STON-R (for S280 N) plus the full-length primers XR-NDE and XR-HIND. S280D and S280 N mutants of KmXR was expressed in E. coli and characterized.
Results and discussion
Cloning and sequence analysis of Kmxyl1 gene from K. marxianus NBRC1777
The K. marxianus genome is currently being sequenced by both the Institut Pasteur (Genolevures) and NITE, but the projects are listed as incomplete [http://genomesonline.org/cgi-bin/GOLD/bin/Search.cgi], thus, the Kmxyl1 gene was cloned using degenerate PCR amplification. The full-length sequence of KmXR (2,036 bp), including the complete ORF (from position 272 to 1,261, total 990 bp), was obtained (GenBank accession No. GU574744) via TAIL-PCR following the method described in section “Cloning of Kmxyl1 gene from K. marxianus NBRC1777”. The potential TATA box in the 5′ region and the polyadenylation site in the 3′ region were found in non-coding regions (Fig. 1). The amino acid sequence of KmXR was highly homologous with other yeast XR (highest was with the XR from K. lactis at 79% identity). All the active sites, substrate-binding residues, and coenzyme utilization were conserved, except for one coenzyme-binding site (Fig. 2) [11, 17].

Alignment of KmXR amino acid sequences with xylose reductases from other yeast species. KmXR, GenBank accession GU574744; PsXR from Pichia stipitis, GenBank accession no. CAA42072, 63% identity; CtXR from Candida tenuis, GenBank accession no. AAC25601, 60% identity; Te-XR: Talaromyces emersonii, GenBank accession no. ACR78268, 55%; KlXR from K. lactis, GenBank accession no. AAA99507.1, 79% identity; KtXR from K. thermotolerans, GenBank accession no. CAR24470.1, 67% identity. The underlined conserved sequences were used to design degenerated primers for the Kmxyl1 cloning. The active site (open circle), substrate-binding residues (open diamond), coenzyme utilization (open inverted triangle) are shown, the Ile-Pro-Lys-Ser motif is enclosed by a rectangle box
Protein expression and purification
After obtaining the full length of gene Kmxyl1, the ORF were amplified by PCR, inserted into pET-21c (Merck, Germany), and subsequently transformed into E. coli BL21 (DE3) for expression. The mutants S280D and S280 N were also expressed in pET-21c by E. coli BL21 (DE3). The recombinant wild-type and mutation KmXRs in the cell lysate were purified by the Ni2+-chelating affinity chromatography and were almost single bands on SDS-PAGE gel, with a molecular mass of around 37 kDa, as calculated theoretically (data not shown).
Characterization of recombinant KmXR
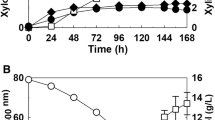
The optimal temperature was studied by measuring XR activity at temperatures ranging from 15–40°C. As shown in Fig. 3a, the optimal temperature was approximately 20°C. To determine the thermal stability, the retained activity of KmXR was measured after incubation for 10 min from 30–50°C. The half-life of KmXR at 42°C was 10 min, whereas the optimal temperature was 20°C (Fig. 3a). The effects of pH on the enzyme activities were determined at pH levels ranging from 5.0–9.0, with the optimal pH at around 6.5 (Fig. 3b).

The activity of XR was determined with 0.2 M xylose and 0.2 mM NADPH. a Thermal profile. The optimal temperature (filled circle) was determined by assaying XR at pH 7.0 with different temperatures, ranging from 15 to 40°C. The thermostability assay (filled square) of XR was taken by treating the enzyme for 10 min at 30–50°C. b Profile of pH effects on XR activity
The effects of metal ions on KmXR activity were also measured. The results showed that Cu2+ and Fe3+ completely inhibited the activity of KmXR. Zn2+ and Ca2+ activated the enzyme greatly and reached 161.06 and 127.10% that of activity without ions, respectively. Other ions had no evident enhancing effects. The 1 mM DTT also improved enzyme activity by approximately 44% (Table 2).
Substrate specificity of KmXR was measured with d-xylose, d-glucose, d-ribose, d-galactose, or d-arabinose as substrates. As shown in Table 3, the optimal substrate was d-xylose (100% activity). Other sugars were converted at 46.92% to 75.77% activity compared with xylose.
XR from K. marxianus NBRC1777 has high coenzyme specificity
The Km and v max values of KmXR for xylose and NADPH were evaluated using the Michaelis–Menten equation. The Km of the K. marxianus XR for d-xylose (33.32 mM) was comparable with those of various XRs, but the Km for NADPH was 65.7 mM, which was higher than those of XRs from other microorganisms. Furthermore, the recombinant KmXR could not use NADH as a coenzyme (Table 4). This result demonstrates that XR from K. marxianus NBRC1777 has high coenzyme specificity.
The K. marxianus is a thermotolerant yeast used in numerous studies on SSFs at elevated temperatures. K. marxianus could not ferment efficiently if xylose, the main product of xylan, is used as the carbon source to produce ethanol. The final concentration of ethanol was only 0.8–5.6 g/l at 35–45°C [2, 4, 34]. Many factors could cause the inefficient xylose fermentation, of which redox imbalance and low enzyme activity in the xylose metabolic pathway are the most likely [7]. In the present study, the XR activity of K. marxianus (1.295 U/mg) was lower than some XRs reported [37, 38], but higher than XR in Debaryomyces hansenii UFV-170 [26, 27]. XDH from K. marxianus NBRC1777 preferred NAD+ as the coenzyme and the activity was very low with NADP+ (data not shown). When the yeast is cultured under oxygen-limited conditions, NADH accumulated and NADPH was depleted. Thus, we deduced that redox imbalance; low activity caused the inefficient xylose fermentation in K. marxianus NBRC1777 under anaerobic culture. The redox imbalance was the key reason for the decreased activity because the yeast would accumulate xylitol during anaerobic culture.
The tetra-amino acid motif Ile276-Pro277-Lys278-Ser279 conserved among NADPH-dependent reductases [17, 18] can be found (enclosed in rectangle box in Fig. 2) in the amino acid sequences of the KmXR proteins. Lys-270 (Lys278 in KmXR) in this motif of Pichia stipitis XR (PsXR) binds to the 2′-phosphate of NADPH [17]. Based on previous mutational studies performed on Candida tenuis XR (CtXR), positions Lys-274, Ser-275, and Asn-276 (Lys-278, Ser-279, and Ser-280 in KmXR; designated by inverted triangles in Fig. 2) emerged as key locations for mutation to increase cofactor specificity for NADH over NADPH (Fig. 2) [17, 18, 24]. The Talaromyces emersonii XRK271R+N273D double mutant has a 16-fold increase in the coenzyme preference for NADH, which shows that lysine and asparagine are the major determinants of NADPH recognition [11]. Additionally, in the PsXR and CtXR, mutations at the same positions greatly increase the preference of XR for NADH [32]. S. cerevisiae that express the K274R/N276D double mutant CtXR and XDH from Galactocandida mastotermitis have 42% increased ethanol yield and 52% decreased xylitol yield [25]. In KmXR, the amino acid residues Lys278-Ser279-Ser280 correspond with the Lys-Ser-Asn of PsXR and CtXR, whereas Asn was substituted with Ser. Ser-280 possibly affected activity and coenzyme preference. Therefore, site-directed mutagenesis was used to check the affects of this site.
Site-directed mutagenesis of the Ser-280 in KmXR
Both S280 N and S280D mutations improved the enzymatic affinity to xylose but greatly reduced their affinity to the coenzyme NADPH (Table 4). The S280 N mutation activity (2.50 U/mg) was almost two times comparing to the wild-type enzyme (1.295 U/mg) and the S280D mutation (0.85 U/mg) was lower than the wild-type. However, the mutants still have no activity with NADH as coenzyme (Table 4). These results indicated that the mutations at this position would reduce the affinity to the NADPH, but they cannot improve the affinity to NADH. The affinity to NADH might be affected by the amino acid residues at other positions.
KmXyl1 disruption inhibits yeast growth on xylose medium
After the Kmxyl1 gene was cloned and the recombinant enzyme was characterized, we needed to confirm whether the cloned Kmxyl1 encoded an XR that catalyzes xylose reduction in the xylose metabolic pathway. After screening the tryptophan dropout SD medium with growth determination the SD medium with xylose (SDX1) or xylitol (SDX2) as sole carbon source, several Kmxyl1 knockout colonies were obtained. Disruption of the Kmxyl1 gene in K. marxianus was confirmed by PCR (Fig. 4a). Using the genomic DNA from the original strain and disrupted strains as template, bands of about 1 and 2.1 kb were amplified, respectively. The 2.1-kb band consisted of the size shift with the trp1 gene insertion and proved that the Kmxyl1 gene in the genome was disrupted (Fig. 4a, lanes 2 and 3). The xylose utilization ability of the Kmxyl1-disrupted strain was determined by measuring growth on the xylose medium. On the SDX1 solid medium, the growth of Kmxyl1-disrupted strains was poorer than those of NBRC1777 and YHJ010, which have the wild-type Kmxyl1 (Fig. 4b). However, on the SDX2 solid medium, all the strains grew well (Fig. 4b). In the SDX1 liquid medium, the difference was more evident: K. marxianus NBRC 1777 and YHJ010 strains grew well and the OD600 reached 1.4 in 25 h, whereas the Kmxyl1–disrupted strains grew weakly (OD600 < 0.1) (Fig. 4c). The result proves that the Kmxyl1 cloned in the present study is responsible for the decreased xylose reduction of K. marxianus NBRC1777.

Disruption of the XR gene Kmxyl1 inhibits yeast growth on xylose medium. a PCR to confirm the disruption of xyl1. M: DNA marker, 1: K. marxianus YHJ010, 2: YZB001, 3: YZB002. b The growth of K. marxianus on SD plate with xylose or xylitol as sole carbon source. c The growth curve of K. marxianus NBRC1777 (filled square), YHJ010 (filled circle) YZB001 (filled triangle) and YZB002 (filled inverted triangle) in liquid SD-xylose medium with xylose as sole carbon source
The disruption of Kmxyl1 caused a deficiency in xylose utilization, but did not hinder xylitol utilization (Fig. 4b). This suggests that KmXR plays an important role in xylose reduction in the xylose metabolic pathway, and only one Kmxyl1 gene in the genome is essential for K. marxianus growth on xylose.
There was insufficient information on the genetic background of K. marxianus, thus, molecular manipulation of the yeast was not performed. The genes, which have the same function as those of other yeast, are often used in K. marxianus research [22]. In the present study, trp1 from S. cerevisiae was used to complement the tryptophan auxotroph of K. marxianus YHJ010 for transformant selection. The compatibility of the selection marker between K. marxianus and S. cerevisiae will enhance research on K. marxianus.
References
Alani E, Cao L, Kleckner N (1987) A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics 116:541–545
Banat IM, Marchant R (1995) Characterization and potential industrial applications of 5 novel, thermotolerant, fermentative, yeast strains. World J Microbiol Biotechnol 11:304–306
Banat IM, Nigam P, Singh D, Marchant R, McHale AP (1998) Ethanol production at elevated temperatures and alcohol concentrations: part I—yeasts in general. World J Microbiol Biotechnol 14:809–821
Banat IM, Singh D, Marchant R (1996) The use of a thermotolerant fermentative Kluyveromyces marxianus IMB3 yeast strain for ethanol production. Acta Biotechnologica 16:215–223
Barron N, Mulholland H, Boyle M, McHale AP (1997) Ethanol production by Kluyveromyces marxianus IMB3 during growth on straw-supplemented whiskey distillery spent wash at 45 degrees C. Bioproc Eng 17:383–386
Bengtsson O, Hahn-Hagerdal B, Gorwa-Grauslund MF (2009) Xylose reductase from Pichia stipitis with altered coenzyme preference improves ethanolic xylose fermentation by recombinant Saccharomyces cerevisiae. Biotechnol Biofuels 2:9
Bera AK, Ho NW, Khan A, Sedlak M (2011) A genetic overhaul of Saccharomyces cerevisiae 424A(LNH-ST) to improve xylose fermentation. J Ind Microbiol Biotechnol 38(5):617–626. doi:10.1007/s10295-010-0806-6
Boyle M, Barron N, McHale AP (1997) Simultaneous saccharification and fermentation of straw to ethanol using the thermotolerant yeast strain Kluyveromyces marxianus IMB3. Biotechnol Lett 19:49–51
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Earley MC, Crouse GF (1996) Selectable cassettes for simplified construction of yeast gene disruption vectors. Gene 169:111–113
Fernandes S, Tuohy MG, Murray PG (2009) Xylose reductase from the thermophilic fungus Talaromyces emersonii: cloning and heterologous expression of the native gene (Texr) and a double mutant (TexrK271R + N273D) with altered coenzyme specificity. J Biosci 34:881–890
Hacker B, Habenicht A, Kiess M, Mattes R (1999) Xylose utilisation: cloning and characterisation of the xylose reductase from Candida tenuis. Biol Chem 380:1395–1403
Hong J, Tamaki H, Akiba S, Yamamoto K, Kumagai H (2001) Cloning of a gene encoding a highly stable endo-beta-1, 4-glucanase from Aspergillus niger and its expression in yeast. J Biosci Bioeng 92:434–441
Hong J, Tamaki H, Kumagai H (2007) Cloning and functional expression of thermostable beta-glucosidase gene from Thermoascus aurantiacus. App Microbiol Biotechnol 73:1331–1339
Hong J, Wang Y, Kumagai H, Tamaki H (2007) Construction of thermotolerant yeast expressing thermostable cellulase genes. J Biotechnol 130:114–123
Jeffries TW (2006) Engineering yeasts for xylose metabolism. Curr Opin Biotechnol 17:320–326
Khoury GA, Fazelinia H, Chin JW, Pantazes RJ, Cirino PC, Maranas CD (2009) Computational design of Candida boidinii xylose reductase for altered cofactor specificity. Protein Sci 18:2125–2138
Lee JK, Koo BS, Kim SY (2003) Cloning and characterization of the xyl1 gene, encoding an NADH-preferring xylose reductase from Candida parapsilosis, and its functional expression in Candida tropicalis. Appl Environ Microbiol 69:6179–6188
Lin Y, Tanaka S (2006) Ethanol fermentation from biomass resources: current state and prospects. Appl Microbiol Biotechnol 69:627–642
Liu YG, Chen Y (2007) High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechniques 43:649–650 652, 654 passim
Margaritis A, Bajpai P (1982) Direct fermentation of d-xylose to ethanol by Kluyveromyces marxianus strains. App Environ Microbiol 44:1039–1041
Nonklang S, Abdel-Banat BM, Cha-aim K, Moonjai N, Hoshida H, Limtong S, Yamada M, Akada R (2008) High-temperature ethanol fermentation and transformation with linear DNA in the thermotolerant yeast Kluyveromyces marxianus DMKU3–1042. Appl Environ Microbiol 74:7514–7521
Nonklang S, Ano A, Abdel-Banat BM, Saito Y, Hoshida H, Akada R (2009) Construction of flocculent Kluyveromyces marxianus strains suitable for high-temperature ethanol fermentation. Biosci Biotechnol Biochem 73:1090–1095
Petschacher B, Leitgeb S, Kavanagh KL, Wilson DK, Nidetzky B (2005) The coenzyme specificity of Candida tenuis xylose reductase (AKR2B5) explored by site-directed mutagenesis and X-ray crystallography. Biochem J 385:75–83
Petschacher B, Nidetzky B (2008) Altering the coenzyme preference of xylose reductase to favor utilization of NADH enhances ethanol yield from xylose in a metabolically engineered strain of Saccharomyces cerevisiae. Microb Cell Fact 7:9
Sampaio FC, de Faria JT, Coimbra JS, Lopes Passos FM, Converti A, Minin LA (2009) Xylose reductase activity in Debaryomyces hansenii UFV-170 cultivated in semi-synthetic medium and cotton husk hemicellulose hydrolyzate. Bioprocess Biosyst Eng 32:747–754
Sampaio FC, de Faria JT, Passos FM, Converti A, Minin LA (2009) Optimal activity and thermostability of xylose reductase from Debaryomyces hansenii UFV-170. J Ind Microbiol Biotechnol 36:293–300
Singh D, Nigam P, Banat IM, Marchant R, McHale AP (1998) Ethanol production at elevated temperatures and alcohol concentrations: Part II—use of Kluyveromyces marxianus IMB3. World J Microbiol Biotechnol 14:823–834
Van Vleet JH, Jeffries TW (2009) Yeast metabolic engineering for hemicellulosic ethanol production. Curr Opin Biotechnol 20:300–306
Verduyn C, Van Kleef R, Frank J, Schreuder H, Van Dijken JP, Scheffers WA (1985) Properties of the NAD(P)H-dependent xylose reductase from the xylose-fermenting yeast Pichia stipitis. Biochem J 226:669–677
Wang XX, Fang BS, Luo JX, Li WJ, Zhang LY (2007) Heterologous expression, purification, and characterization of xylose reductase from Candida shehatae. Biotechnol Lett 29:1409–1412
Watanabe S, Pack SP, Saleh AA, Annaluru N, Kodaki T, Makino K (2007) The positive effect of the decreased NADPH-preferring activity of xylose reductase from Pichia stipitis on ethanol production using xylose-fermenting recombinant Saccharomyces cerevisiae. Biosci Biotechnol Biochem 71:1365–1369
Watanabe S, Saleh AA, Pack SP, Annaluru N, Kodaki T, Makino K (2007) Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein engineered NADP+ -dependent xylitol dehydrogenase. J Biotechnol 130:316–319
Wilkins MR, Mueller M, Eichling S, Banat IM (2008) Fermentation of xylose by the thermotolerant yeast strains Kluyveromyces marxianus IMB2, IMB4, and IMB5 under anaerobic conditions. Proc Biochem 43:346–350
Woodyer R, Simurdiak M, van der Donk WA, Zhao H (2005) Heterologous expression, purification, and characterization of a highly active xylose reductase from Neurospora crassa. Appl Environ Microbiol 71:1642–1647
Yokoyama SI, Suzuki T, Kawai K, Horitsu H, Takamizawa K (1995) Purification, characterization and structure-analysis of nadph-dependent d-xylose reductases from Candida-tropicalis. J Ferment Bioeng 79:217–223
Zeng QK, Du HL, Wang JF, Wei DQ, Wang XN, Li YX, Lin Y (2009) Reversal of coenzyme specificity and improvement of catalytic efficiency of Pichia stipitis xylose reductase by rational site-directed mutagenesis. Biotechnol Lett 31:1025–1029
Zhang F, Qiao D, Xu H, Liao C, Li S, Cao Y (2009) Cloning, expression, and characterization of xylose reductase with higher activity from Candida tropicalis. J Microbiol 47:351–357
Acknowledgments
We thank Professor Tamaki Hisanori from Kagoshima University and Professor Kumagai Hidehiko from Ishikawa Prefectural University for providing the K. marxianus YHJ010 and plasmid YEGAP. This work was supported by a grant-in-aid from the National Natural Science Foundation of China (Grant no. 31070028) and the project was also sponsored by National Basic Research Program of China (Grant No. 2011CBA00801), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, and the Specialized Research Fund for the Doctoral Program of Higher Education of China (Grant no. 20093402120027).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, B., Zhang, L., Wang, D. et al. Identification of a xylose reductase gene in the xylose metabolic pathway of Kluyveromyces marxianus NBRC1777. J Ind Microbiol Biotechnol 38, 2001–2010 (2011). https://doi.org/10.1007/s10295-011-0990-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-011-0990-z