
Abstract
Microbiological synthesis of higher alcohols (1-butanol, isobutanol, 2-methyl-1-butanol, etc.) from plant biomass is critically important due to their advantages over ethanol as a motor fuel. In recent years, the use of branched-chain amino acid (BCAA) biosynthesis pathways together with heterologous Ehrlich pathway enzyme system (Hazelwood et al. in Appl Environ Microbiol 74:2259–2266, 2008) has been proposed by the Liao group as an alternative approach to aerobic production of higher alcohols as new-generation biofuels (Atsumi et al. in Nature 451:86–90, 2008; Atsumi et al. in Appl Microbiol Biotechnol 85:651–657, 2010; Cann and Liao in Appl Microbiol Biotechnol 81:89–98, 2008; Connor and Liao in Appl Environ Microbiol 74:5769–5775, 2008; Shen and Liao in Metab Eng 10:312–320, 2008; Yan and Liao in J Ind Microbiol Biotechnol 36:471–479, 2009). On the basis of these remarkable investigations, we re-engineered Escherichia coli valine-producing strain H-81, which possess overexpressed ilvGMED operon, for the aerobic conversion of sugar into isobutanol. To redirect valine biosynthesis to the production of alcohol, we also—as has been demonstrated previously (Atsumi et al. in Nature 451:86–90, 2008; Atsumi et al. in Appl Microbiol Biotechnol 85:651–657, 2010; Cann and Liao in Appl Microbiol Biotechnol 81:89–98, 2008; Connor and Liao in Appl Environ Microbiol 74:5769–5775, 2008; Shen and Liao in Metab Eng 10:312–320, 2008; Yan and Liao in J Ind Microbiol Biotechnol 36:471–479, 2009)—used enzymes of Ehrlich pathway. In particular, in our study, the following heterologous proteins were exploited: branched-chain 2-keto acid decarboxylase (BCKAD) encoded by the kdcA gene from Lactococcus lactis with rare codons substituted, and alcohol dehydrogenase (ADH) encoded by the ADH2 gene from Saccharomyces cerevisiae. We show that expression of both of these genes in the valine-producing strain H-81 results in accumulation of isobutanol instead of valine. Expression of BCKAD alone also resulted in isobutanol accumulation in the culture broth, supporting earlier obtained data (Atsumi et al. in Appl Microbiol Biotechnol 85:651–657, 2010) that native ADHs of E. coli are also capable of isobutanol production. Thus, in this work, isobutanol synthesis by E. coli was achieved using enzymes similar to but somewhat different from those previously used.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Production of biofuel from renewable sources is of particular interest due to the limited reserves of traditional energy sources (such as fossil fuels), unstable oil prices, and the need to limit greenhouse gas emissions and air and water pollution. Plant biomass is one of the most important renewable energy resources. Significant steps have been taken to increase biomass use to produce hexoses and pentoses and to adapt microorganisms for the simultaneous consumption of a variety of carbon sources. Conversion of C5 and C6 sugars into liquid energy carriers occurs in two major anaerobic biological processes: ethanol fermentation and mixed acetone, ethanol and butanol fermentation. Currently, biosynthesis of higher alcohols (1-butanol, isobutanol, 2-methyl-1-butanol, etc.) from plant biomass has gained significant attention due to advantages over ethanol as a motor fuel. In particular, isobutanol has higher energy density than ethanol, and it is less hygroscopic and less volatile. Up until now, among higher alcohols, only 1-butanol has been manufactured by microbiological synthesis, and this was achieved by using Clostridium acetobutylicum in a strictly anaerobic process [16, 18]. Microorganisms naturally capable of industrially relevant conversion of sugars into C4 and C5 alcohols have not yet been identified. The yeast Saccharomyces cerevisiae has been shown to synthesize small amounts of isobutanol and other higher alcohols by the Ehrlich pathway as result of catabolism of corresponding branched-chain amino acids (BCAA) valine, leucine, and isoleucine [10–12, 22].
Recently, a strategy for aerobic production of the higher alcohols has been proposed [2, 3, 5, 7, 23, 27]. This approach includes using the highly active BCAA biosynthetic pathways as a source of keto acids (2-ketoisovalerate, 2-keto-3-methylvalerate, 2-ketoisocaproate, 2-ketovalerate, etc.) and subsequent conversion of these metabolites into the corresponding alcohols (isobutanol, 2-methyl-1-butanol, 3-methyl-1-butanol, 1-butanol, etc.) [2, 3, 5, 7, 23, 27]. In the cited works [2, 3, 27], to produce isobutanol, the conversion of carbon source into 2-ketoisovalerate in Escherichia coli was achieved, through an overexpression of ilvCD (E. coli) genes together with enhanced expression of genes encoding different types of acetolactate synthase, ilvIH (E. coli) or alsS (Bacillus suibtilis).
BCAAs are usually manufactured by fermentation by employing mutant strains of various bacteria, including E. coli. As the immediate precursors for BCAAs can also be used for the biosynthesis of higher alcohols, an opportunity to adapt the existing BCAA-hyperproducing strains for biofuel production is of particular interest. Overall in this work, redesign of valine producer H-81 for isobutanol synthesis was performed. In this producer, 2-ketoisovalerate supply for isobutanol production was ensured by overexpression of ilvD and ilvGM genes encoding feedback-resistant acetolactate synthase II of E. coli. Critical to isobutanol production strategy are the following two enzymes from the Ehrlich pathway: 2-keto acid decarboxylase (EC 4.1.1.72) and alcohol dehydrogenase (EC 1.1.1.1), with broad substrate specificity [2, 3, 5, 7, 23, 27]. Particularly, 2-keto acid decarboxylase Kivd from Lactococcus lactis [8], together with alcohol dehydrogenase Adh2 from S. cerevisiae, has been used for isobutanol synthesis in E. coli [2, 3, 27]. Considering substrate specificity, another well-characterized 2-keto acid decarboxylase from L. lactis, KdcA, seems to be also applicable for the same purpose [24, 28]. The substrate spectrum of KdcA is similar to Kivd, with which it shares approximately 90% sequence identity [8, 24, 28].
In this study, by introducing the kdcA gene from L. lactis, with rare codons substituted, and the ADH2 gene from S. cerevisiae under the control of a regulated promoter into an E. coli valine-producing strain, we evaluated the possibility of producer re-engineering for aerobic conversion of sugar to higher alcohols, particularly isobutanol.
Materials and methods
Strains and media
All strains used in this study are listed in Table 1. XL1-Blue was the E. coli strain used for cloning. Strain H-81 was derived from E. coli MG1655 (ilvG ValR ileS17) and contains four copies of the valine operon under the control of the PR-promoter of phage λ, PR-ilvG ValR MED-2Tfd, integrated into the chromosome via mini-Mu vector [21, 26]. E. coli strains were grown aerobically at 37°C in either lysogeny broth (LB) medium [20] or M9 medium [20] containing 100 μg/ml of ampicillin for selection, if appropriate. Cultivation of valine-producing strains was carried out in fermentation medium (FM) at pH 7.0 containing the following (g/l): (NH4)2SO4 15; KH2PO4 1.5; MgSO4 1; CaCO3 20; B1 0.01; glucose 60. S. cerevisiae S288C strain was grown aerobically at 37°C in yeast peptone dextrose (YPD) medium [1].
Cultivation conditions
Preseeding cultures in test tubes containing 1 ml of LB medium were incubated at 32°C for 18 h on a rotary shaker (250 rpm). Overnight cultures were diluted 1:20 into 2 ml of FM medium in 20 × 200-mm test tubes by adding 0.5 mM isopropyl-thio-α-galactoside (IPTG) for Lac promoter induction. Cultivation of strain H-81 and its plasmid derivatives was performed at 32°C for 68 h on a rotary shaker (250 rpm).
Determination of amino acid concentration
Accumulated L-valine was measured by thin-layer chromatography (TLC). TLC plates (10 × 15) cm were coated with 0.11-mm layers of Sorbfil silica gel containing no fluorescent indicator (Stock Company Sorbpolymer, Krasnodar, Russia). The Sorbfil plates were developed with a mobile phase consisting of isopropanol-ethylacetate-25% aqueous ammonia-water (16:16:5:10 v/v). A solution of ninhydrin (2% w/v) in acetone was used as the visualizing reagent.
Reagents
Restriction enzymes, T4 DNA ligase, and high-fidelity polymerase chain reaction (PCR) enzyme mix were obtained from Fermentas (Vilnuis, Lithuania). QIAquick Gel Extraction Kits and QIAGEN Plasmid Mini Kits were obtained from QIAGEN GmbH (Germany). Oligonucleotides were obtained from Sintol (Moscow, Russia). Schiff’s reagent for aldehyde determination was from Fluka. Isobutanal, 2-methylbutanal, isobutanol and 2-methylbutanol were from Sigma.
DNA handling procedures
Protocols for genetic manipulation of S. cerevisiae [1] and E. coli [20], as well as techniques for isolating and manipulating nucleic acids were as described. Plasmid DNA was isolated with a QIAGEN Plasmid Mini Kit and genomic DNA was isolated with a Bacterial Genomic DNA Kit (Sigma). DNA fragments were isolated from agarose gels using a QIAquick Gel Extraction Kit (QIAGEN). Synthesis of the kdcA gene was performed by Sloning BioTechnology GmbH (Germany).
Plasmid construction
All plasmids used or constructed in this study are listed in Table 1.
1. Construction of pPCRScript-kdcA-L.la: To clone kdcA, synthesis of a kdcA gene (GenBank accession number AY548760) from L. lactis with a modified nucleotide sequence codon-optimized for E. coli and with EcoRI and BamHI restriction sites (5′-GAATTCGAAGGAGGGAATGTG-3′, 5′-GGATCCTTA-3′) was carried out by Sloning BioTechnology. In the modified kdcA sequence, the following 52 codons were optimized: 16 Thr codons—positions 2, 70, 113, 130, 137, 142, 185, 186, 188, 231, 237, 287, 291, 371, 385, and 459 (codon ACA was replaced with ACG); 8 Arg codons—positions 10, 133, 147, 156, 320, 421, 443, and 462 (codons CGA, CGG and AGA were replaced with CGC); 14 Pro codons—positions 23, 91, 99, 128, 158, 164, 175, 207, 250, 348, 352, 398, 408, and 448 (codons CCA, CCT, and CCC were replaced with CCG); 9 Leu—114, 153, 233, 238, 276, 302, 358, 440, and 523 (codon CTA was replaced with CTG); and 5 Ile—255, 304, 308, 332, and 453 (codon ATA was replaced with ATT). The resulting DNA fragment containing the kdcA gene was digested with EcoRI and BamHI and cloned into pPCR-Script (GenBank accession number U46017.1) cut with the same enzymes, creating pPCRScript-kdcA-Lla. The kdcA gene was cloned in the opposite orientation relative to the Lac promoter to reduce potential toxicity from gene expression.
2. Construction of pMW118-kdcA: To clone kdcA, pPCRScript-kdcA-Lla was digested with EcoRI and BamHI. The DNA fragment containing the kdcA gene was cloned into the pMW118 plasmid cut with the same enzymes, creating pMW118-kdcA.
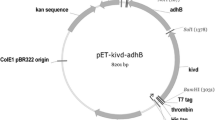
3. Construction of pMW118-kdcA-adh2: The structural parts of the ADH2 and ADH1 genes from S. cerevisiae S288C have nearly 100% homology; therefore, cloning the ADH2 gene was done by two-step PCR amplification using two pairs of primers (Fig. 1). Initially, to amplify the 1,520-bp DNA fragment containing the ADH2 gene, S. cerevisiae S288C genomic DNA was used as a PCR template with primers adh2-L1 (5′-GACAAAAAAAATGATGGAAGACAC-3′) and adh2-R1 (5′-CAGGAAAGAGTTACTCAAGAAC-3′). PCR products were purified and used as a PCR template to amplify the ADH2 gene with primers adh2-L (5′-CTGACTGGATCCAAGAAGGAGATATATATATGTCTATCCCAGAAACTCAAAAAG -3′) and adh2-R (5′-GCCATAGTCGACTTATTTAGAAGTGTCAACAACGTAT-3′). PCR products were digested with BamHI and SalI and cloned into pMW118-kdcA cut with the same enzymes, creating pMW118-kdcA-adh2 (Fig. 2).

The two-step polymerase chain reaction (PCR) amplification scheme for the ADH2 gene from Saccharomyces cerevisiae S288C genomic DNA

Map of the plasmid pMW118-kdcA-adh2
Preparation of cell extracts
A culture of strain H-81 harboring either pMW118-kdcA or pMW118-kdcA-adh2 was grown overnight, and 0.1 ml was used to inoculate fresh medium (10 ml of M9 medium supplemented with 1/10 v/v LB). The cells were induced with 0.5 mM IPTG after inoculation. Inoculated cultures were grown for 4.5 h until an optical density at 540 nm of 0.8 was reached. Cells were harvested by centrifugation, washed twice in 1M sodium chloride and then in 0.1 M potassium–phosphate buffer (pH 6.3). Cells were suspended in 0.1 M potassium–phosphate buffer (pH 6.3) and disrupted by sonication. The supernatant after centrifugation at 12,000× for 30 min was used as the cell extract. All steps were performed at temperatures <4°C. Protein concentrations were determined by the Bradford assay (Bio-Rad protein assay, GmbH).
BCKAD assay
For branched-chain 2-keto acid decarboxylase (BCKAD) assays, cell extracts were incubated for 3 h at 30°C in a closed 2-ml vial at pH 6.3 (0.1 M potassium–phosphate buffer) with 0.1 M 2-ketoisovalerate (2-keto-3-methylvalerate) as the substrate and with 1 mM MgCl2 and 0.1 mM thiamine pyrophosphate (TPP) as cofactors in a total volume of 1 ml. Then, 0.2 ml of Schiff’s reagent was added into the reaction mixture to determine aldehyde concentration. After color development, samples were placed on ice. The concentrations of isobutanal and 2-methyl-butanal were determined spectrophotometrically at 553 nm using a spectrophotometer (Spectronic GENESYS 2, Thermo Electron Corporation). Relative 2-keto acid decarboxylase activity was calculated by dividing the amount of product formed (in nanomoles) by the reaction time (in minutes) and the protein content in the sample (in milligrams).
Branched-chain amino acid transaminase assay
Cell extracts were incubated for 15 min at 37°C in 2-ml vial at pH 7.5 [0.5 M Tris hydrochloride (HCl) buffer + 1 mM dithiothreitol (DTT)] with 50 mM Phe and 50 mM 2-ketoisovalerate (2-keto-3-methylvalerate) as the substrates and with 0.5 mM pyridoxal phosphate (PLP) as cofactor in a total volume of 200 μl. Then, the samples were placed on ice and 0.8 ml of 1.25 N NaOH was added to stop the reaction. The formation of phenylpyruvate was analyzed at 320 nm against a control to which NaOH had been added at 0 min. The OD was read immediately after addition of NaOH. A molar extinction coefficient of 17,500 M−1 cm−1 was used [6]. Specific activity is defined as nanomoles of phenylpyruvate formed per minute per milligram of protein.
Determination of isobutanol concentration in FM medium
The concentration of isobutanol in FM medium was determined by gas chromatography (GC) analysis. GC flame ionization detector (GC-FID) analysis was accomplished on a Varian 3600 gas chromatograph with 3 m × 3-mm (i.d.) stainless-steel column packed with 15% CARBOWAX 1500 on CHROMATON N-AW 0.15–0.2 mm. The injector temperature was 180°C, and injection volume was 1 μl. Nitrogen was used as the carrier gas with a flow rate of 30 ml/min. The column temperature was initially kept at 50°C for 3 min and then increased to 120°C at 10°C/min and held for 3 min. The detector temperature was 250°C. Methanol, ethanol, 1-propanol, isobutanol, and 1-butanol were separated under these conditions.
Results and discussion
The valine-producing strain H-81 [21, 26] was used as a model host for isobutanol production from glucose. This strain converts glucose into valine mainly due to the expression of ilvGMED genes under control of the “strong” lambda phage PR promoter [19]. Additionally, this artificial operon (PR-ilvGMED) contains the full-length gene ilvG. Due to this mutation, strain H-81 (in contrast to IlvG-deficient wild-type E. coli K-12) possesses a type II acetolactate synthase that is resistant to feedback inhibition by valine. This strain accumulated 12–13 g/l of valine from 60 g/l of glucose in test-tube cultivation (Table 2), thereby indicating the potential for equimolar synthesis of 2-ketoisovalerate. The latter compound can act not only as a keto precursor of valine but also as an isobutanol precursor (Fig. 3).

Pathway of isobutanol biosynthesis. IlvGM acetolactate synthase II, IlvC acetohydroxy acid isomeroreductase, IlvD dihydroxy-acid dehydratase, IlvE branched-chain amino acid aminotransferase, Kdc 2-keto acid decarboxylase, Adh alcohol dehydrogenase
E. coli does not possess an enzyme capable of decarboxylating branched-chain keto acids (BCKAs). The possibility to use for this purpose heterologous kivd gene (GenBank Accession Number AJ746364) from L. lactis [8] was shown by Liao group [2, 3, 27]. In our work, we used the kdcA gene (GenBank accession number AY548760) from L. lactis [24], which shares almost 90% sequence identity with the kivd gene L. lactis [28]. The kdcA gene from L. lactis encodes a BCKA decarboxylase (EC 4.1.1.72) with broad substrate specificity [24, 28]. This enzyme enables conversion of BCKAs (2-ketoisovalerate, 2-keto-3-methylvalerate and 2-ketoisocaproate), which are the immediate keto precursors of valine, isoleucine, and leucine into the corresponding aldehydes (isobutanal, 2-methylbutanal and 3-methylbutanal, respectively) [24, 28]. The native nucleotide sequence of kdcA contains a number of codons that are rare in E. coli. These codons were optimized for expression in E. coli and are listed in “Materials and methods.” Synthesis of the modified kdcA sequence was performed by Sloning Biotechnology GmbH. The kdcA-containing EcoRI-BamHI DNA fragment was cloned into a high-copy-number pPCR-Script vector. The resulting plasmid (pPCRScript-kdcA-Lla) contains the kdcA coding region oriented away from the lactose promoter to avoid possible toxicity from kdcA gene expression.
The kdcA gene with rare codons substituted was subcloned into the low-copy-number vector pMW118. The resulting plasmid (pMW118-kdcA) contains the kdcA gene under the control of the P lac promoter. Introduction of this plasmid into the valine-producing H-81 strain and subsequent induction with IPTG resulted in a drop in valine production accompanied by low-level isobutanol accumulation during cultivation in test tubes under aerobic conditions (Table 2). Using a rubber stopper for cultivation of isobutanol-accumulating strains prevented alcohol evaporation and thereby increased isobutanol accumulation in culture broth (Table 2). No detectable isobutanol synthesis was observed in the absence of pMW118-kdcA (Table 2). Induction of kdcA expression initiated 2-ketoisovalerate decarboxylation, thereby decreasing valine production and yielding isobutanal. It is worth noting that the further conversion of isobutanal into the corresponding alcohol, isobutanol, is apparently due to the native E. coli alcohol dehydrogenases’ ability to carry out this reaction [3]. The E. coli genome includes at least the following six genes annotated as alcohol dehydrogenases (ADHs) or putative ADHs: adhE, adhP, eutG, yiaY, yqhD and yjgB [3, 14] (www.ecocyc.org). Higher alcohols containing more than three carbon atoms in the chain have been identified as preferred substrates for yqhD-encoded ADH [25]. Our data concerning isobutanol production by the valine-producing strain under conditions of kdcA expression are also in good agreement with the recent observation that the YqhD protein can act as an isobutanol dehydrogenase [3].
In addition to the native E. coli putative isobutanol dehydrogenases, a range of heterologous enzymes can be used for more effective conversion of isobutanal into isobutanol. A wide range of microorganisms possess this kind of activity. In particular, the yeast S. cerevisiae is responsible for degradation of BCAAs into fusel alcohols via the Ehrlich pathway [12, 13]. The S. cerevisiae genome harbors genes for a range of ADHs, which are carefully reviewed in [9, 13], some of which can participate in fusel alcohols biosynthesis. Five ethanol dehydrogenases, Adh1, Adh2, Adh3, Adh4, Adh5, and two nicotinamide adenine dinucleotide phosphate, reduced (NADPH)-dependent medium-chain alcohol dehydrogenases, Adh6 and Adh7, with broad substrate specificity are among them [9, 13]. From among these enzymes, we chose to use Adh2 for heterologous expression in E. coli. It is necessary to note that quite recently, due to careful investigation by Atsumi et al. [3], AdhA L. lactis and YqhD E. coli were established as preferable enzymes for isobutanol synthesis [3]. The S. cerevisiae isozymes Adh1 and Adh2 are practically identical except for one amino acid residue; Adh1 contains Met at position 294, whereas Adh2 contains Leu at this position. Nucleotide sequence analysis revealed the presence of several codons that are rare in E. coli in the structural part of ADH2 (8 Arg codons encoded by AGA and 3 Leu codons encoded by CTA), but none were located in the beginning of the gene; therefore ADH2 was cloned in its native form. The DNA fragment containing ADH2 was obtained by two-step PCR amplification (as shown in Fig. 1) by using the genomic DNA of S. cerevisiae S288C as a template for the initial step. This fragment was subcloned into the plasmid pMW118-kdcA just downstream of kdcA. The resulting plasmid, pMW118-kdcA-adh2, contains an artificial inducible operon, P lac -kdcA-ADH2. The level of both valine and isobutanol accumulation during test tube cultivation of the recombinant strain H-81/pMW118-kdcA-adh2 under IPTG induction was tested (Table 3). Data show that induction of the operon decreased valine synthesis to a level sixfold less than that of the control parent strain, H-81. The drop in valine synthesis was accompanied by isobutanol accumulation. Despite the variation in valine accumulation with and without induction, isobutanol production did not differ greatly between these two cases. Evidently, this fact indicated that a considerable part of the newly synthesized isobutanol evaporated during aerobic cultivation. The absence of a marked drop in biomass due to toxic levels of isobutanol after IPTG induction supports this idea. Aerobic incubation of test tubes with cell-free medium initially containing 10 g/l of isobutanol resulted in the evaporation of >80% of the initial alcohol (the final concentration of isobutanol after incubation was <2 g/l). Thus, our data indicate that expression of two heterologous genes encoding BCKAD and ADH in E. coli redirected the carbon flux from the synthesis of valine to that of isobutanol; but for all tested aerobic conditions, the yield of isobutanol was 1.5% (g isobutanol/g glucose). To accumulate higher levels of isobutanol in the culture broth, it is necessary to perform cultivation under special conditions that prevent evaporation. In this case, when microaerobic conditions arise, it is necessary to disrupt a number of genes coding for pyruvate-degrading enzymes that are activated anaerobically, as was suggested by Liao and coworkers [2].
Measurement of BCKAD activity in recombinant strains containing the kdcA gene from L. lactis under control of the P lac promoter confirmed the presence of IPTG-inducible decarboxylase activity (Table 4). Also, it is important to note that the enzymatic activity toward 2-keto-3-methylvalerate, the keto precursor of isoleucine, was 1.5 times higher than the activity toward 2-ketoisovalerate, the keto precursor of valine (Table 4). These data agree with the results of detail analysis of substrate specificity of KdcA L. lactis performed by Yep and coworkers [28].
Expression of the P lac -kdcA-ADH2 operon did not completely prevent valine oversynthesis (Table 3). Apparently there exists competition between the IlvE-mediated amination of 2-ketoisovalerate and utilization of this keto acid by BCKAD and further conversion by ADH. The level of IlvE activity for H-81 was 340 nmol/min/mg. It is necessary to take into account that the host strain H-81 contains several chromosomal copies of the ilvGMED operon under control of the “strong” lambda phage PR promoter, whereas the kdcA and ADH2 genes are in an artificial operon under control of the “moderate” P lac promoter. The moderate promoter was chosen as a first approach to prevent possible accumulation of toxic aldehyde in case of imbalance between its synthesis and further consumption. Therefore, imbalance between effective 2-ketoisovalerate synthesis and its further insufficient utilization in the Ehrlich pathway seems quite possible, thereby indicating the need to optimize expression of the kdcA-ADH2 artificial operon.
Conclusions
This work highlights the possibility of using BCAA biosynthetic pathways for biofuel production from renewable resources. Using as a model the E. coli valine-producing strain with the overexpressed genes encoding acetolactate synthase II, the aerobic synthesis of isobutanol from glucose was achieved by coupling BCAA biosynthetic pathways in the host with heterologous Ehrlich pathway, which completely support the data first obtained by Liao et al. [2, 3, 27]. It was shown that the valine-producing E. coli strain H-81 can be re-engineered for isobutanol production by introducing an artificial operon consisting of two heterologous genes: kdcA from L. lactis and ADH2 from S. cerevisiae. Also, the ability of E. coli to convert isobutanal into isobutanol by a native ADH was confirmed, and YqhD is considered to be one of the promising candidates. To further improve isobutanol production, the balance between 2-keto acid synthesis and utilization should be maintained by optimizing kdcA and ilvE gene expression. A high level of ilvE expression is not needed. Toxicity of higher alcohols remains a difficult problem. Isobutanol tolerance of E. coli was studied in detail [4]. Only few reports have addressed the potential of alternative hosts for alcohol production and focused on the selection of microorganisms tolerant to these compounds [15, 17]. Considering the significant recent progress in the development of membrane technologies for higher alcohol separation and the promising results of screening for and characterizing alcohol-tolerant species, further engineering of microorganisms for effective conversion of biomass-derived sugars into fusel alcohols should create a basis for implementing this approach to the production of biotechnological fuels.
References
Amberg D, Burke D, Strathern J (2005) Methods in yeast genetics, 2005th edn. Cold Spring Harbor Laboratory Course Manual, Cold Spring Harbor, NY
Atsumi S, Hanai T, Liao JC (2008) Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451:86–90. doi:10.1038/nature06450
Atsumi S, Wu TY, Eckl EM, Hawkins SD, Buelter T, Liao JC (2010) Engineering the isobutanol biosynthetic pathway in Escherichia coli by comparison of three aldehyde reductase/alcohol dehydrogenase genes. Appl Microbiol Biotechnol 85:651–657. doi:10.1007/s00253-009-2085-6
Brynildsen MP, Liao JC (2009) An integrated network approach identifies the isobutanol response network of Escherichia coli. Mol Sys Biol 5:277. doi:10.1038/msb.2009.34
Cann AF, Liao JC (2008) Production of 2-methyl-1-butanol in engineered Escherichia coli. Appl Microbiol Biotechnol 81:89–98. doi:10.1007/s00253-008-1631-y
Collier RH, Kohlhaw G (1972) Nonidentity of the aspartate and the aromatic aminotransferase components of transaminase A in Escherichia coli. J Bacteriol 112:365–371
Connor MR, Liao JC (2008) Engineering of an Escherichia coli strain for the production of 3-methyl-1-butanol. Appl Environ Microbiol 74:5769–5775. doi:10.1128/AEM.00468-08
de la Plaza M, Fernandez de Palencia P, Pelaez C, Requena T (2004) Biochemical and molecular characterization of alphaketoisovalerate decarboxylase, an enzyme involved in the formation of aldehydes from amino acids by Lactococcus lactis. FEMS Microbiol Lett 238:367–374. doi:10.1016/j.femsle.2004.07.057
de Smidt O, du Preez JC, Albertyn J (2008) The alcohol dehydrogenases of Saccharomyces cerevisiae: a comprehensive review. FEMS Yeast Res 8:967–978. doi:10.1111/j.1567-1364.2008.00387
Dickinson JR, Harrison SJ, Hewlins MJE (1998) An investigation of the metabolism of valine to isobutyl alcohol in Saccharomyces cerevisiae. J Biol Chem 273:25751–25756. doi:10.1074/jbc.273.40.25751
Dickinson JR, Harrison SJ, Dickinson JA, Hewlins MJE (2000) An investigation of the metabolism of isoleucine to active amyl alcohol in Saccharomyces cerevisiae. J Biol Chem 275:10937–10942. doi:10.1074/jbc.275.15.10937
Dickinson JR, Salgado LEJ, Hewlins MJE (2003) The catabolism of amino acids to long chain and complex alcohols in Saccharomyces cerevisiae. J Biol Chem 278:8028–8034. doi:10.1074/jbc.M211914200
Hazelwood LA, Daran JM, van Maris AJA, Pronk JT, Dickinson JR (2008) The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Appl Environ Microbiol 74:2259–2266. doi:10.1128/AEM.02625-07
Keseler IM, Bonavides-Martinez C, Collado-Vides J, Gama-Castro S, Gunsalus RP, Johnson DA, Krummenacker M, Nolan LM, Paley S, Paulsen IT, Peralta-Gil M, Santos-Zavaleta A, Shearer AG, Karp PD (2009) EcoCyc: A comprehensive view of Escherichia coli biology. Nucleic Acids Res 37:D464–D470. doi:10.1093/nar/gkn751
Knoshaug EP, Zhang M (2009) Butanol tolerance in a selection of microorganisms. Appl Biochem Biotechnol 153:13–20. doi:10.1007/s12010-008-8460-4
Lin YL, Blaschek HP (1983) Butanol production by a butanol-tolerant strain of Clostridium acetobutylicum in extruded corn broth. Appl Environ Microbiol 45:966–973
Luo LH, Seo PS, Seo JW, Heo SY, Kim DH, Kim CH (2009) Improved ethanol tolerance in Escherichia coli by changing the cellular fatty acids composition through genetic manipulation. Biotechnol Lett 31:1867–1871. doi:10.1007/s10529-009-0092-4
Nair RV, Bennett GN, Papoutsakis ET (1994) Molecular characterization of an aldehyde/alcohol dehydrogenase gene from Clostridium acetobutylicum ATCC 824. J Bacteriol 176:871–885
Ptashne M (1986) A genetic switch: gene control and phage λ. Cell Press & Blackwell Scientific, Oxford
Sambrook J, Russell D (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Savrasova EA (2007) Development of the mini-Mu system for highly effective integration and amplification of genetic material into the chromosome of E. coli without selective marker. Ph.D. Thesis, Moscow
Sentheshanuganathan S (1960) The mechanism of the formation of higher alcohols from amino acids by Saccharomyces cerevisiae. Biochem J 74:568–576
Shen CR, Liao JC (2008) Metabolic engineering of Escherichia coli for 1-butanol and 1-propanol production via the keto-acid pathways. Metab Eng 10:312–320. doi:10.1016/j.ymben.2008.08.001
Smit BA, van Hylckama Vlieg JET, Engels WJM, Meijer L, Wouters JTM, Smit G (2005) Identification, cloning, and characterization of a Lactococcus lactis branched-chain α-keto acid decarboxylase involved in flavor formation. Appl Environ Microbiol 71:303–311. doi:10.1128/AEM.71.1.303-311.2005
Sulzenbacher G, Alvarez K, Van Den Heuvel RH, Versluis C, Spinelli S, Campanacci V, Valencia C, Cambillau C, Eklund H, Tegoni M (2004) Crystal structure of E. coli alcohol dehydrogenase YqhD: evidence of a covalently modified NADP coenzyme. J Mol Biol 342:489–502. doi:10.1016/j.jmb.2004.07.034
Tabolina E, Rybak K, Khourges E, Voroshilova E, Gusyatiner M (2005) Method for producing L-amino acid using bacteria belonging to the genus Escherichia. US Patent 7476531
Yan Y, Liao JC (2009) Engineering metabolic systems for production of advanced fuels. J Ind Microbiol Biotechnol 36:471–479. doi:10.1007/s10295-009-0532-0
Yep A, Kenyon GL, McLeish MJ (2006) Determinants of substrate specificity in KdcA, a thiamin diphosphate-dependent decarboxylase. Bioorg Chem 34:325–336. doi:10.1016/j.bioorg.2006.08.005
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Savrasova, E.A., Kivero, A.D., Shakulov, R.S. et al. Use of the valine biosynthetic pathway to convert glucose into isobutanol. J Ind Microbiol Biotechnol 38, 1287–1294 (2011). https://doi.org/10.1007/s10295-010-0907-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-010-0907-2