
Abstract
Background
Benzyl acetate is an aromatic ester with a jasmine scent. It was discovered in plants and has broad applications in food, cosmetic, and pharmaceutical industries. Its current production predominantly relies on chemical synthesis. In this study, Escherichia coli was engineered to produce benzyl acetate.
Results
Two biosynthetic routes based on the CoA-dependent β-oxidation pathway were constructed in E. coli for benzyl acetate production. In route I, benzoic acid pathway was extended to produce benzyl alcohol by combining carboxylic acid reductase and endogenous dehydrogenases and/or aldo-keto reductases in E. coli. Benzyl alcohol was then condensed with acetyl-CoA by the alcohol acetyltransferase ATF1 from yeast to form benzyl acetate. In route II, a plant CoA-dependent β-oxidation pathway via benzoyl-CoA was assessed for benzyl alcohol and benzyl acetate production in E. coli. The overexpression of the phosphotransacetylase from Clostridium kluyveri (CkPta) further improved benzyl acetate production in E. coli. Two-phase extractive fermentation in situ was adopted and optimized for benzyl acetate production in a shake flask. The most optimal strain produced 3.0 ± 0.2 g/L benzyl acetate in 48 h by shake-flask fermentation.
Conclusions
We were able to establish the whole pathway for benzyl acetate based on the CoA-dependent β-oxidation in single strain for the first time. The highest titer for benzyl acetate produced from glucose by E. coli is reported. Moreover, cinnamyl acetate production as an unwanted by-product was very low. Results provided novel information regarding the engineering benzyl acetate production in microorganisms.
Graphical abstract

Similar content being viewed by others
Introduction
Benzyl acetate is an aromatic ester that has attracted attention for its scent that is similar to jasmine, gardenia, and lily of the valley. It is commonly used as a base fragrance ingredient in perfumes, cosmetics, and soap fragrances; it is also one of the largest varieties in the synthetic fragrance industry [1]. It is found naturally in cloves, chamomile, jasmine, and hyacinth; however, extracts obtained from these plants are insufficient to meet the global usage (about 10,000 tons per year). Therefore, benzyl acetate is mainly produced via chemical synthesis. Among chemical synthesis methods, the esterification reaction of acetic acid and benzyl alcohol (BALC) is the most commonly used. It is generally performed in the presence of H2SO4, but it causes disadvantages such as equipment corrosion, excessive side reactions, and severe environmental pollution. As a precursor, BALC is conventionally produced and commercially prepared from petroleum. This process involves benzyl chloride via alkaline hydrolysis [2]. In addition to harsh reaction conditions, non-renewable feedstocks are used in this process, thus posing sustainability issues.
For a more sustainable approach, the precursor BALC was synthesized from glucose through the systematic engineering of Escherichia coli [3]. An artificial pathway is constructed, starting from phenylpyruvate, which is the direct precursor of phenylalanine (L-Phe). This pathway is composed of three enzymes, namely, hydroxymandelate synthase (HmaS) from Amycolatopsis orientalis, mandelate dehydrogenase (MdlB), and phenylglyoxylate decarboxylase (MdlC) from Pseudomonas putida. These three enzymes sequentially convert phenylpyruvate to benzaldehyde (BALD) via mandelate and phenylglyoxylate. Multiple endogenous alcohol dehydrogenases (ADHs) and aldo-keto reductases (AKRs) in E. coli convert BALD into BALC. This route involves two decarboxylation reactions and is therefore less carbon efficient [3].
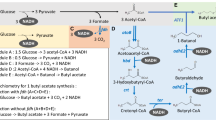
The CoA-dependent β-oxidation pathway via the common precursor L-Phe releases acetyl-CoA, fueling the TCA cycle or supplying the substrate for the synthesis of an acetylated product, and this process is more carbon efficient. In the pathway, phenylalanine ammonia lyase converted L-Phe to cinnamic acid (CA), and cinnamate: CoA ligase catalyzed CA to cinnamyl-Coenzyme A (CA-CoA). Enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase, and 3-oxoacyl-CoA ketohydrolase sequentially transformed CA-CoA to BA via 3-hydroxyphenylpropionyl-CoA (HPP-CoA), 3-ketophenylpropionyl-CoA (KPP-CoA) [4, 5] (Fig. 1, Route I). Basing on this pathway, Otto et al. engineered benzoic acid (BA) production in Pseudomonas taiwanensis, which produced approximately 232.0 and 366.4 mg/L of BA from glucose and glycerol, respectively [4]. In another study, the CoA-dependent β-oxidative pathway was evaluated for BA production in E. coli, and the resulting strain produced 2.37 g/L BA from glucose [6]. In principle, when a carboxylic acid reductase (CAR) [7] is expressed simultaneously, BA produced through in this route could be convert BALD [6] (Fig. 1, Route I). And then endogenous ADHs and AKRs in E. coli can spontaneously convert BALD to BALC. In plants, BALD is a native product. Different from the above pathway, 3-ketoacyl-CoA thiolase (KAT) converts KPP-CoAin the β-oxidative route into benzoyl-CoA (BA-CoA) in plants, and BA-CoA is reduced to BALD by benzaldehyde synthase (BS) (Fig. 1, Route II) [8].
Benzyl acetate is synthesized with lipase in the presence of BALC by biocatalytic approach [9, 10]. Studies have also described microbial conversion for benzyl acetate biosynthesis via external BALC addition. For example, approximately 150 mg/L of benzyl acetate can be produced with 2 g/L BALC by expressing a thermostable chloramphenicol acetyltransferase from Staphylococcus aureus (CATSa) in E. coli [11]. When the engineered E. coli carrying an alcohol acyltransferase (ATF1) from Saccharomyces cerevisiae is supplemented with 2.0 g/L BALC, 1177.98 ± 45.72 mg/L of benzyl acetate can be produced [12].
De novo biosynthesis of benzyl acetate using a heterologous host has been rarely attempted except for the following nice study published very recently. Choi et al. reported a strategy for the efficient production of benzyl acetate in E. coli [13]. Specially, they designed a BA-dependent pathway based on the CoA-dependent β-oxidation route, which contained upstream module converting glucose to BA and downstream module transforming BA into benzyl acetate. After various trials, two modules could not be integrated into single strain. Thus, a co-culture strategy was applied: (1) Firstly, the Bn1 strain containing upstream module is inoculated alone to produce BA. (2) After 48 h, the Bn-BnAc3 strain containing downstream module is added to the bioreactor to convert the BA to benzyl acetate. In an optimized two-phase extractive fermentation process, 2238.3 ± 171.9 mg/L of benzyl acetate from glucose in 108 h was produced by the co-culture of two strains, and the by-product cinnamyl acetate reached 797.8 ± 220.9 mg/L [13].
Herein, we report our efforts that led to the high-level production of benzyl acetate in E. coli by one-step simple fermentation. Two different biosynthetic routes based on the CoA-dependent β-oxidative pathway were constructed in E. coli strains for benzyl acetate production (Fig. 1). In route I, firstly, a CAR specific activity toward BA was characterized, and thus we were able to achieve BALC production in E. coli strain by integrating biosynthetic pathway of BA and the CAR. Benzyl acetate production was achieved and enhanced by screening acetyltransferases with better catalytic properties. In route II, in order to avoid using CAR, a plant CoA-dependent β-oxidation pathway via benzoyl-CoA was assessed for BALC and benzyl acetate production in E. coli. Finally, production of benzyl acetate was enhanced by overexpressing CkPta to elevate the pool of acetyl-CoA. Two-phase in situ extractive fermentation was adopted and optimized by using different organic solvents and ratios. Ultimately, the best-performing engineered single strain produced 3.0 ± 0.2 g/L benzyl acetate from simple carbon resource in shake-flask at 48 h and the by-product cinnamyl acetate product was only 2.89 ± 0.29 mg/L.

De novo biosynthesis of benzyl acetate in engineered E. coli strain via the carboxylic acid reductase pathway (route I) and the benzaldehyde synthase pathway (route II). Abbreviations: CA, trans-cinnamic acid; CA-CoA, cinnamoyl-CoA; HPP-CoA, 3-hydroxyphenylpropionyl-CoA; KPP-CoA, 3-ketophenylpropionyl-CoA; BA-CoA, benzoyl-CoA; BA, benzoic acid; BALD, benzaldehyde; BALC, benzyl alcohol; Ac-CoA, acetyl-CoA; E4P, erythrose 4- phosphate; PEP, phosphoenolpyruvate; DHAP, 3-deoxy-D-arabinoheptulosonate 7-phosphate; SHK, shikimate; CHA, chorismate; PRE, prephenate; 4HPPA, 4-hydroxyphenylpyruvate; L-Tyr, L-tyrosine; PPA, phenylpyruvate; L-Phe, L-phenylalanine; PYR, pyruvate; TCA, tricarboxylic acid cycle; AcP, acetyl- phosphate. Enzymes involved in pathway: PykA, PYR kinase II; PykF, PYR kinase I; CkPta, Phosphotransacetylase from C. kluyveri; AroGfbr, feedback resistant mutants of DHAP synthase AroG; PheAfbr, feedback-resistant mutant of PRE dehydratase; TyrR, aromatic amino acid biosynthesis and transport regulon transcriptional regulator. TyrA, PRE dehydrogenase; RgPAL, phenylalanine ammonia-lyase; ScCCLA294G, cinnamate: CoA ligase; PhdE, enoyl-CoA hydratase; PhdB, 3-hydroxyacyl-CoA dehydrogenase; PhdC, 3-oxoacyl-CoA ketohydrolase (acetyl-CoA forming); CAR, carboxylic acid reductase; PPTase, phosphopantetheinyl transferase; KAT, 3-ketoacyl thiolase; PhBS, benzaldehyde synthase; ADHs, endogenous alcohol dehydrogenases; AKRs, aldo-keto reductases; AATs, alcohol acyl transferases. Solid arrows represent single reactions, dashed arrows represent multiple reactions, and dashed lines with T end represents feedback inhibition. The red X indicates gene deletion. Endogenous genes are shown in black. The genes overexpressed are highlighted in blue. The differences between route (I) and route (II) are irrespectively shown in light pink and light orange
Results and discussion
Establishment of L-Phe over-producing chassis strains
L-Phe is an important intermediate in benzyl acetate biosynthesis routes (Fig. 1). To obtain L-Phe over-producing chassis strains, we chose to further engineer the U7 strain constructed in our lab [14]. First, pheAfbr encoding feedback-resistant prephenate dehydratase was controlled by a strong constitutive Pcore-trc promoter [15] and integrated into the aromatic amino acid exporter yddG locus of strain U7 to restore the L-Phe production and decrease L-Phe export, resulting in the U7A strain. Then aroGfbr encoding feedback-resistant 3-deoxy-D-arabinoheptulosonate 7-phosphate synthase (DHAP) was controlled by the Pcore-trc promoter and integrated into the yghX locus of U7A to enhance the metabolic flux toward the shikimate pathway, generating the U7AG strain. Then we deleted tyrA of U7AG to block the tyrosine biosynthetic pathway, forming the GAP strain. 0.5 mL of overnight seed culture of U7A, U7AG, and GAP was irrespectively inoculated into 50 mL of LB medium in 250 mL shake flasks and cultivated at 37 °C with shaking at 220 rpm. After 48 h of culturing, the fermentation broth containing cells was ultra-sonicated and centrifuged. The titers of L-Phe in supernatant were detected via 2, 4-dinitrofluorobenzene derivation methods and HPLC analysis. As results shown in Fig. S1, the L-Phe production of strain U7AG (0.95 ± 0.04 g/L) was twice as high as that of strain U7A (0.45 ± 0.01 g/L). The aroGfbr overexpression under the control of Pcore-trc effectively enhanced L-Phe production. The GAP strain had the highest L-Phe production at 1.02 ± 0.03 g/L. Therefore, it was used as a chassis strain to construct the biosynthetic pathway of benzyl acetate.
Benzyl alcohol biosynthesis by extending CoA-dependent β-oxidative pathway of benzoic acid biosynthesis with carboxyl acid reductase in E. coli
Luo et al. biosynthesized BA from L-Phe based on the CoA-dependent β-oxidative pathway I-variant [6]. In this work, we designed a route for BALC biosynthesis from glucose via BA as shown in Fig. 1. The pivotal step in this route was the reduction of BA to BALD catalyzed by CARs. The aldehyde molecule can be spontaneously converted to alcohols by native ADHs and AKRs in E. coli. CARs usually are promiscuous to many carboxylic acid substrates [7, 16]. CA as the main intermediate can also be converted to cinnamyl alcohol with CARs; consequently, CA cannot be converted to a CoA derivative. Therefore, a CAR with high substrate specificity toward BA should be obtained to successfully construct the pathway. In this study, eight CARs, namely, PcCAR4 from Pycnoporus cinnabarinus [17], NPS11 from Serpula lacrymans [18], MavCAR from Mycobacterium avium, MmCAR from Mycobacterium marinum, MsCAR from Mycobacterium smegmatis, SeCAR from Segniliparaceae rotundus, SrCAR from Segniliparus rugosus, and NiCAR from Nocardia Iowensis [14] were tested for their activity against BA via heterologous expression in E. coli and feeding experiments with BA to verify our hypothesis. PcCAR4 has a low substrate specificity, and BA is its preferred substrate [17]. NPS11 has a 10-fold higher preference for BA than for CA [18]. These CARs were expressed under the T7 promoter control in E. coli BL21 (DE3). In feeding experiments with BA or CA, the corresponding BALC or cinnamyl alcohol was detected (Table S3). PcCAR4 could reduce BA with a conversion rate of about 24.3% ± 1.5%, and no cinnamyl alcohol was detected. Thus, PcCAR4 showed distinct BA substrate specificity. NPS11 could use BA as a substrate with a 3.6% conversion rate and could not convert CA into the corresponding product. PcCAR4 and NPS11 were selected as the candidates for constructing the BALC biosynthetic pathway (route I). MavCAR, MmCAR, MsCAR, SeCAR, and NiCAR exhibited high catalytic activities for CA and BA with approximately 80% substrate conversion rate and did not show a clear BA preference. SrCAR showed a 95.0% ± 8.1% BA conversion rate as indicated by BALC production and a 54.9% ± 3.7% CA conversion rate as demonstrated by cinnamyl alcohol production.
Starting from L-Phe for BA biosynthesis, the CoA-dependent β-oxidative pathway I-variant has been reported (Fig. 1). Phenylalanine ammonia lyase (RgPAL) converted L-Phe to CA, and cinnamate: CoA ligase (ScCCLA294G) catalyzed CA to CA-CoA. Enoyl-CoA hydratase (PhdE), 3-hydroxyacyl-CoA dehydrogenase (PhdB), and 3-oxoacyl-CoA ketohydrolase (PhdC) were sequentially recruited to convert CA-CoA to BA [6]. RgPAL, ScCCLA294G, PhdE, PhdB, and PhdC were cloned into pRSFDuet-1 expressed under the T7 promoter control, resulting in pRSF-I (Fig. 2A). pET-NPSsfp harboring NPS11 and pET-Pcppa harboring PcCAR4 were constructed (Fig. 2A). Furthermore, pET-NPSsfp or pET-Pcppa was introduced to the L-Phe-producing strain GAP with pRSF-I, respectively producing EcBC01 and EcBC02 strains, which were cultured in the G medium. BALC produced by EcBC01 and EcBC02 was detected via HPLC and LC–HRMS analyses (Fig. 2B). The cell growth of EcBC01 was nearly equal to that of EcBC02. The EcBC02 strain produced 302.9 ± 28.3 mg/L BALC, which was 3.7 times higher than that of BALC produced by the EcBC01 strain (79.7 ± 4.5 mg/L). The results suggested that PcCAR4 had a higher efficiency for BALC synthesis than NPS11. In addition, 699.0 ± 44.0 mg/L BA and 132.9 ± 8.4 mg/L CA accumulated in the fermentation broth of the EcBC02 strain (Fig. 2C), and no peak of cinnamyl alcohol was detected.
MavCAR, MmCAR, MsCAR, SeCAR, NiCAR, and SrCAR showed good catalytic activities for CA and BA. Thus, they were unsuitable for constructing the biosynthetic pathway of BALC. However, considering that SrCAR exhibited BA preference over CA, we incorporated SrCAR into the BALC synthetic pathway in E. coli. We co-transformed pRSF-I and pET-SrCARsfp carrying SrCAR into the GAP strain, resulting in the EcBC-Sr strain. Almost no BALC was detected in the EcBC-Sr strain, and 469.9 ± 6.4 mg/L cinnamyl alcohol accumulated as the major product (Fig. S2).

Biosynthesis of BALC from glucose via the route I or route II. (A) Schematic illustration of the constructed plasmids for overexpressing genes encoding RgPAL, ScCCLA294G, PhdE, PhdB, PhdC, PcCAR4, PPTase, NPS11, Sfp, KAT1 and PhBS. PT7, T7 promoter. (B) The BALC production of recombinant strains. EcBC01 harboring pRSF-I and pET-NPSsfp, EcBC02 harboring pRSF-I and pET-Pcppa, EcBC03 strain harboring pRSF-II and pET-BS. (C) The HPLC analysis of the metabolites produced by strains EcBC02 and the control strain containing plasmids of pETDuet-1 and pRSFDuet-1. Values and error bars represent the means and standard deviations of biological triplicates
Selection of acetyltransferases for the de novo biosynthesis of benzyl acetate in E. coli
After BALC was synthesized in E. coli, acetyl-CoA/BALC acetyltransferases (AATs) with higher activities should be identified. ATT candidates from different resources were tested for their ability to produce benzyl acetate from BALC. ATF1 from S. cerevisiae S288C with unselective various alcohols [19], CATec3-Y20F, an engineered chloramphenicol acetyltransferase (CAT) emerging as a promising enzyme for aromatic and terpene esters production [20, 21], and ANN09798 from Nicotiana tabacum [22] were examined for benzyl acetate synthesis. The genes encoding ATF1, CATec3-Y20F, and ANN09798 were introduced to the EcBC02 strain, yielding the strains EcYZ01, EcYZ02, and EcYZ03, respectively. These strains were fermented using a two-phase in situ extraction strategy with a 1:10 (v/v) dodecane overlay to avoid ester volatilization in the shake flasks. The metabolites in the aqueous and organic phases were analyzed with HPLC. The results were shown in Fig. 3. A peak corresponding to benzyl acetate (tR = 17.2 min) was observed in the HPLC chromatogram of the metabolites in the dodecane phase and produced by all three recombinant strains (Fig. 3A). The results were further confirmed via GC–MS analysis (Fig. 3B). Thus, benzyl acetate could be biosynthesized from glucose in the E. coli recombinant strain. Most of the produced benzyl acetate was extracted into the dodecane organic phase, and only a small amount of benzyl acetate was left in the aqueous phase (Fig. 3C, Table S4). The total benzyl acetate production of EcYZ01 strain harboring ATF1, EcYZ02 strain harboring CATec3-Y20F, and EcYZ03 strain carrying ANN09798 was 1151.8 ± 35.4, 810.7 ± 81.6, and 530.1 ± 38.5 mg/L, respectively. Thus, ATF1 was more suitable than CATec3-Y20F and ANN09798 for benzyl acetate synthesis (Fig. 3D). Benzyl acetate total production of EcYZ01 with a 1:10 (v/v) dodecane overlay was four times higher than that without a dodecane overlay (Fig. 3D). The two-phase in situ extractive fermentation was highly effective in improving benzyl acetate production. The by-product cinnamyl acetate was detected in dodecane phase of the three strains culture (Fig. S3), and its production in EcYZ01 harboring ATF1 was 0.35 ± 0.02 mg/L, which was far lower than that in EcYZ02 and EcYZ03. We detected the CA accumulated (Fig. 3C) and did not observe the peak of cinnamyl alcohol in fermentation broth of three strains. It suggested that PcCAR4 could utilize CA to synthesize cinnamyl alcohol in strains harboring a de novo biosynthetic pathway of benzyl acetate. The strain EcYZ04 (GAP containing pRSF-I and pET-NPSsfp-ATF1) only produced 32.78 ± 5.5 mg/L benzyl acetate ((Fig. 3D).

Benzyl acetate produced by recombinant strains. (A)The HPLC analysis of metabolites in dodecane phase, produced by the EcYZ01 (pRSF-I and pET-Pcppa-ATF1), EcYZ02 (pRSF-I and pET-Pcppa-CAT) and EcYZ03 (pRSF-I and pET-Pcppa-798). (B) The GC-MS analysis of benzyl acetate produced by recombinant strains and benzyl acetate standard. (C) The HPLC analysis of metabolites in the organic and aqueous phases produced by the strain EcYZ01. The control strain, GAP containing empty plasmids(pETDuet-1 and pRSFDuet-1). The metabolites’ location is represented by a vertical dashed line. (D) The total production of benzyl acetate of EcYZ01, EcYZ02, EcYZ03, EcYZ04 and EcYZ05. EcYZ04 harboring pRSF-I and pET-NPSsfp-ATF1, and EcYZ05 harboring pRSF-II and pET-BS-ATF1 using the two-phase in situ extractive fermentation with 1:10 (v/v) dodecane overlay. EcYZ01 (no), fermentation of EcYZ01 strain without dodecane overlay. Values and error bars represent the means and standard deviations of duplicate experiments
Synthesis of benzyl acetate by the heterologous expression of plant CoA-dependent β-oxidative pathway of benzaldehyde biosynthesis and ATF1 (route II) in E. coli
In this work, we attempted to circumvent the problem of the promiscuous activity of CARs for the synthesis of precursor BALC in route I by recruiting the plant CoA-dependent β-oxidative pathway in route II (Fig. 1). Route II has a common intermediate KPP-CoA. KAT converts KPP-CoA into BA-CoA by β-oxidative shortening of the propyl side-chain of KPP-CoA. Huang (2022) identified the heterodimeric benzaldehyde synthase PhBS, which is composed of α and β subunits and is responsible for BALD formation from BA-CoA in plants [8]. PhBS exhibits strict substrate specificity toward BA-CoA and uses NADPH as a cofactor. PhBS and PhKAT1 derived from Petunia hybrida [23] were recruited to establish the pathway from KPP-CoA to BALD. pRSF-II carrying ScCCLA294G, RgPAL, PhdB, and PhKAT1 was constructed for BA-CoA biosynthesis (Fig. 1). pRSF-II was simultaneously introduced to GAP together with pET-BS harboring PhBS, generating the EcBC03 strain. The strain was subjected to fermentation for the analysis of BALC production. The recombinant EcBC03 strain produced 283.0 ± 24.6 mg/L BALC (Fig. 2B), which was slightly lower than that produced by EcBC02 harboring genes involved in route I (302.9 ± 28.3 mg/L). Subsequently, the gene encoding ATF1 was incorporated into the EcBC03 strain, generating the EcYZ05 strain. EcYZ05 produced 122.3 ± 14.3 mg/L benzyl acetate in two-phase in situ extractive fermentation with a dodecane overlay of 1:10 (v/v) culture volume (Fig. 3D). The amount of benzyl acetate produced via route I was nine times higher than that of via route II.
Optimization of benzyl acetate production by precursor and cofactor enhancement
NADPH is a key cofactor in the reduction steps in both routes. Pos5p encoding NADH kinase from S. cerevisiae could directly phosphorylate NADH to produce NADPH. The overexpression of Pos5p* (Pos5p without the mitochondrial targeting sequence) in E. coli effectively enhances the productivity of NADPH-dependent bioprocesses [24]. To reinforce NADPH regeneration improve BA reduction efficiency and further enhance benzyl acetate biosynthesis, we integrated Pos5p* under the T7 promoter control into the aslA locus of GAP strain, resulting in GAP01 strain. The plasmids of pRSF-I and pET-Pcppa-ATF1 were cotransformed into GAP01, creating the EcYZ06 strain. As results shown in Fig. 4A, the cell growth of EcYZ06 was similar to that of EcYZ01 (GAP containing pRSF-I and pET-Pcppa-ATF1), and the total production of benzyl acetate in EcYZ06 was 1039.7 ± 29.8 mg/L, which was slightly lower than in EcYZ01 (1151.8 ± 35.4 mg/L) by two-phase in situ extractive fermentation with a dodecane overlay of 1:10 (v/v) culture volume. Thus, the NADPH supply might not be a limiting factor.
CkPta, a phosphotransacetylase converting acetyl phosphate into acetyl-CoA, has been applied in microbial hosts for efficient synthesis of many products using acetyl-CoA as a precursor [25, 26]. The codon-optimized CkPta was controlled by the Pcore-trc promoter and integrated into the E. coli native Pta locus of GAP strain, generating the GAP01 strain. The plasmids of pRSF-I and pET-Pcppa-ATF1 were co-transformed into GAP01, creating the EcYZ07 strain. Benzyl acetate production of EcYZ07 strain was evaluated by two-phase in situ extractive fermentation with a dodecane overlay of 1:10 (v/v) culture volume. The total production of benzyl acetate in EcYZ07 was 1532.3 ± 31.5 mg/L, which showed a 30% increase compared with the strain EcYZ01 (1151.8 ± 35.4 mg/L). The cell growth of EcYZ07 (OD600 = 15.2) was 1.15 times that of EcYZ01 (Fig. 4A). The results demonstrated the overexpression of CkPta in E. coli-native Pta locus effectively improved benzyl acetate production in our engineered strain. We surmised overexpression of CkPta elevated the acetyl-CoA pool in the engineered strain.

Optimization of benzyl acetate production. (A) The total titers of benzyl acetate in different chassis strains containing pRSF-I and pET-Pcppa-ATF1. EcYZ01 (derived from GAP chassis strain), EcYZ06 (derived from GAP01 chassis strain with Pos5P* overexpression), EcYZ07 (derived from GAP02 chassis strain with CkPta overexpression). (B) The cell growth and the total benzyl acetate production of EcYZ07 strain with different ratios (v/v) of dodecane overlay in two-phase in situ extractive fermentation. (C) The cell growth and the total benzyl acetate production of EcYZ07 strain with 1:1 (v/v) ratio of olive oil and isopropyl myristate overlay in two-phase in situ extractive fermentation
Improvement of benzyl acetate production via the optimization of two-phase in situ extractive fermentation
Two-phase in situ extractive fermentation helps relieve product toxicity and improve the final product titer. Esters can be readily secreted outside of cells and easily separated from the culture broth via dual-phase extractive fermentation [27]. In the present work, a two-phase in situ extraction strategy was used to produce benzyl acetate in shake-flask fermentation. Initially, 1:10 (v/v) dodecane was used as the extractive agent to enhance benzyl acetate accumulation, as mentioned above. Two ratios, namely, 1:1 (v/v) and 1:3 (v/v), were evaluated for fermentation to further optimize the extraction system. The best-performing strain, EcYZ07, for benzyl acetate production was used to evaluate the in-situ extraction agent. Cell growth and benzyl acetate in the aqueous and organic phases were detected (Fig. 4B, Table S5). With 1:1 (v/v) dodecane overlay, benzyl acetate in the organic phase reached 2460.7 ± 199.6 mg/L, and approximately 57.5 ± 4.4 mg/L benzyl acetate was left in the aqueous phase (Table S5). The total production of benzyl acetate with a 1:1 (v/v) dodecane overlay was improved by 1.6 times that of a 1:10 (v/v) dodecane overlay. The cell growth under the dodecane overlay at three different ratios with a similar OD600 of approximately 14 was nearly 1.5 times as that without a dodecane overlay.
Olive oil and isopropyl myristate are usually selected as nutritious and safe solvents in nutraceutical, pharmaceutical, and cosmetic products [28, 29]. We examined their effects on benzyl acetate production when they were used as extractive organic agents at 1:1 (v/v). The amounts of benzyl acetate production of overlays with olive oil and isopropyl myristate were 1407.6 ± 47.0 or 2459.3 ± 179.7 mg/L, respectively (Fig. 4C, Table S6). No benzyl acetate could be detected in the aqueous phase with isopropyl myristate as the overlay. The improvement of benzyl acetate production of overlay with isopropyl myristate was almost equal to that of dodecane at the same ratio of 1:1 (v/v). However, a certain amount of intermediate CA was also extracted in the organic phase (Fig. S4). An ideal solvent should have high selectivity for the extracted compound [30], thus, 1:1 (v/v) dodecane in situ extractive fermentation was the optimal solvent for improving benzyl acetate synthesis.
The time profiles of cell growth and the titers of the main metabolites produced by the strain EcYZ07 with a 1:1 (v/v) dodecane overlay fermentation were investigated. The samples were withdrawn every 12 h after overnight seed cultures were inoculated into the G medium. As shown in Fig. 5A, cell growth entered the stationary phase, and OD600 was maintained at 14–16 after 48 h of fermentation. After 48 h of fermentation, benzyl acetate production reached 3.0 ± 0.2 g/L and gradually stabilized. Other metabolites were also detected. As shown in Fig. 5B, C, the by-product cinnamyl acetate production only was 2.89 ± 0.29 mg/L, there was no BA accumulated, but 6.7 ± 0.4 mg/L CA accumulated after 48 h of fermentation.

The two-phase in situ extractive fermentation of the engineered E. coli EcYZ07 strain (GAP02 containing pRSF-I and pET-Pcppa-ATF1) with 1:1 (v/v) dodecane overlay in shake flasks. (A) The profiles of benzyl acetate total production and growth. (B) The profiles of CA and BA production. (C) The HPLC analysis of metabolites produced by EcYZ07 in aqueous and dodecane phases. Experiments were performed in triplicate; error bars represent standard deviations
Conclusion
There is great interest in the development of glucose as a sustainable resource for the biomanufacturing of benzyl acetate. We report for the first time, a single-host microbial process for benzyl acetate production from glucose. Two biosynthetic pathways based on CoA-dependent β-oxidation were assessed for the de novo production of benzyl acetate. Seven enzymes were recruited to extend the biosynthetic pathway of benzyl acetate from L-Phe in E. coli. Through pathway enzymes screening, elevating acetyl-CoA pool and optimizing two-phase extractive fermentation, the corresponding engineered strain achieved a highest titer of 3.0 ± 0.2 g/L in shake-flask at 48 h.
Methods and materials
Strains, media, and chemicals
E. coli DH5α was used as a cloning host in plasmid construction. E. coli BL21 (DE3) was utilized to express genes. E. coli K-12 MG1655 derivative strain U7 [14] was used as the base strain. U7 derivatives and plasmids are listed in Table 1. E. coli strain was routinely cultured in a Luria–Bertani (LB) medium or on LB agar plates. The M9Y medium containing 20.0 g/L glucose, 0.25 g/L yeast extract, 3.0 g/L KH2PO4, 1 g/L NH4Cl, 17.1 g/L Na2HPO4·12H2O, 0.5 g/L NaCl, 14.7 mg/L CaCl2·H2O, and 246.5 mg/L MgSO4·7H2O was used in feeding experiments. A G medium with the following components was used for de novo production (per liter): 10.0 g/L glycerol, 6.8 g/L KH2PO4, 17.9 g/L Na2HPO4·12H2O, 0.71 g/L Na2SO4, 2.67 g/L NH4Cl, 15.0 g/L tryptone, 5.0 g/L yeast extract, 2 mM MgSO4, and 0.1 mM CaCl2. MgSO4 and CaCl2 were added from the respective stock solutions, which were prepared and autoclaved separately. Ampicillin (100 mg/L) or kanamycin (50 mg/L) was added appropriately. Standards containing BALC, BA, CA, cinnamyl alcohol, cinnamyl acetate, and benzyl acetate were purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China).
Plasmid construction
All plasmids used or constructed in this work are listed in Table 1. The oligonucleotide primers used for plasmid construction are listed in Table S1. Codon-optimized DNA sequences in E. coli were chemically synthesized by GENEWIZ (Table S2). Gibson assembly and molecular cloning techniques were used in plasmid construction.
The CARs-sfp fragments of MavCAR-sfp, MmCAR-sfp, MsCAR-sfp, SeCAR-sfp, SrCAR-sfp, NiCAR-sfp PCR-amplified from the corresponding plasmids of pETDuet-amS-CAR-sfp [14] (stocked in our lab) by using the primers CARmav-F/CARmm2-F2/CARms-F/CARni-F/CARsr-F/CARse-F and CARsr-R. The plasmids of pET-MavCARsfp, pET-MmCARsfp, pET-MsCARsfp, pET-SeCARsfp, pET-SrCARsfp, and pET-NiCARsfp were assembled with CAR-sfp and pETDuet-1 fragments amplified using pET (CAR)-F and pET (CAR)-R primers.
ScCCLA294G from Streptomyces coelicolor A3(2) [31] was synthesized and amplified using NdeI-ScCCL and XhoI-ScCCL primers; then, it was cloned into the pRSFDuet-1 plasmid via NdeΙ and BglΙI double digestion to create the pRSF-CL plasmid. RgPAL from Rhodotorula glutinis JN-1 [32] (GenBank: AUQ35650.1) was synthesized and cloned into pRSFDuet-CL between NcoI and BamHI to create the pRSF-Rg-CL plasmid. PhdBC and PhdE were amplified from Corynebacterium glutamicum ATCC 13,032 [33] by using the following respective primers to construct pET-BCE: NdeI-phdBC and phdBC-3R; XhoI-phdE and phdE-5 F. Overlapping PCRs were performed to obtain the PhdBCE fragment. Then, the PhdBCE gene fragment was cloned into pETDuet-1 via the double restriction sites of NdeΙ and BglII to generate pET-BCE. The EcoRI-phdBCE-HindIII gene fragment was amplified from pET-BCE by using the primers EcoRI-phdBCE and HindIII-phdBCE and cloned into pRSF-Rg-CL between the restriction sites of EcoRI and HindIII to generate pRSF-I.
CARs require posttranslational modification by an auxiliary phosphopantetheinyl transferase (PPTase) in order to obtain full enzymatic activity. The gene encoding phosphopantetheinyl transferase in E. coli (EcPPTase) was amplified from E. coli MG1655 by using the primers EcPPTase-5 F and EcPPTase-R2; afterward, it was cloned into pETDuet-1 between the restriction sites of NcoI and HindIII to generate pET-ppa. PcCAR4 from P. cinnabarinus (GenBank: OM908756.1) was synthesized and amplified using the primers PcCAR-5 F and PcCAR-3R; then, it was cloned into pET-ppa via the restriction sites of NdeΙ and XhoI to create pET-Pcppa. NPS11 from S. lacrymans (GenBank: KX118591.1) was synthesized and amplified using the primers NPS11-F2 and NPS11-R2. Subsequently, it was cloned into pETDuet-1 harboring sfp encoding phosphopantetheinyl transferase from Bacillus subtilis (GenBank: WP_015715234.1) between the restriction sites of EcoRI and NdeI to create pET-NPSsfp. ATF1 (GenBank: NC_001147.6) was synthesized and amplified using ATF1-9 F and ATF1-9R, and then, was assembled into pET-NPSsfp to generate pET-NPSsfp-ATF1. ATF1 was amplified with ATF1-6 F and ATF1-6R. Two fragments were amplified using PPase-6R/PPase-6R and V-F/PcCAR-3R with pET-Pcppa as the template and assembled using ClonExpress II one-step cloning kit (Vazyme) with the ATF1 fragment, yielding pET-Pcppa-ATF1. Similarly, the engineered CATec3-Y20F from C. thermocellum (UniProtKB/Swiss-Prot: P00484.1) and ANN09798 from N. tabacum (GenBank: AF500202.1) were synthesized and amplified using the primers CAT-6 F/CAT-6R and 798-F/798-R, respectively; subsequently, they were separately assembled into pET-Pcppa to generate pET-Pcppa-CAT and pET-Pcppa-798.The PhKAT1 fragment (GenBank: FJ657663.1) was synthesized and amplified using the primers phKAT1-5 F and phKAT1-3R. Afterward, it was assembled with the pRSF-Rg-CL-BE fragment amplified from pRSF-I by using phdE-5 F and phdB-3R in ClonExpress II one-step cloning kit (Vazyme), yielding pRSF-II. PhBS, including PhBSα (GenBank: OK095279) and PhBSβ (GenBank: OK095280), from P. hybrida was synthesized. PhBS (PhBSα–PhBSβ) was subcloned into pETDuet-1 with a maltose-binding protein (MBP) tag by using ClonExpress II one-step cloning kit (Vazyme), yielding pET-BS. ATF1 was amplified using ATF1 (BS)-F and ATF1 (BS)-R and then assembled into the pET-BS fragment to construct pET-BS-ATF1.
Construction of strains
The CRISPR-Cas9 system was used to efficiently manipulate the genome of the engineered E. coli. The U7 strain was used as a starting strain, as shown in Table 1. The CRISPR-Cas9 system was were employe to genome editing. To knock out tyrA, the pCas9 carrying relevant N20 sequence and homologous arms was introduced to the U7 strain to locate and cleave target genes. The pheAfbr [34] under the Pcore-trc promoter [15] control was integrated into yddG locus. The feedback-resistant mutant aroGfbr (AroGD146N) under the Pcore-trc promoter was integrated into the yghX locus. Pos5P* from S. cerevisiae under the T7 promoter control was integrated into the aslA locus (Pos5P*, Pos5P with 17 amino acid from the N-terminal sequence being truncated). CkPta (Gene bank: WP_012101779) under the Pcore-trc promoter control was integrated into the E. coil native Pta (EcoPta) site.
Feeding experiments
The plasmids of pET-MavCARsfp, pET-MmCARsfp, pET-MsCARsfp, pET-SeCARsfp, pET-SrCARsfp, pET-NiCARsfp, pET-NPSsfp, and pET-Pcppa were individually transferred into E. coli BL21 (DE3). A single colony of the transformant was inoculated into 5 mL of LB medium with ampicillin and cultivated at 37 °C overnight with shaking at 200 rpm. The overnight culture was transferred into 50 mL of fresh LB medium with ampicillin and cultivated at 37 °C. When OD600 reached 0.6–0.8, a final concentration of 0.1 mM isopropyl-β-D-thiogalactoside (IPTG) was added to the shake flasks and then cultivated at 16 °C for 16 h. The cells were harvested through centrifugation and suspended in 50 mL of M9Y medium. Then, the CA or BA substrate was added to the cultures at a final concentration of 2 mM. After 48 h of cultivation at 30 °C and 220 rpm, the supernatant was sampled and analyzed through HPLC. Data were collected from three independent experiments.
Shake-flask culture conditions
A fresh colony of the recombinant E. coli strain was inoculated into 5 mL of LB medium and cultivated at 37 °C overnight with shaking at 220 rpm. Then, 0.5 mL of overnight seed culture was inoculated into 45 mL of G medium in 250 mL shake flasks and cultivated at 37 °C with shaking at 220 rpm. When OD600 reached 0.8–1.0 (in about 3 h), a final concentration of 0.1 mM IPTG was added. After 16 h of induction at 23 °C and 220 rpm, 5 mL of 20% glucose was added to shake flasks. Cultivation was continued at 30 °C for 48 h with shaking at 220 rpm. BALC and benzyl acetate were detected in the broth.
The two-phase in situ extractive fermentation was conducted as follows. A fresh colony of the recombinant E. coli strain was inoculated into 5 mL of LB medium and cultivated at 37 °C overnight with shaking at 220 rpm. Then, 0.5 mL of overnight seed culture was inoculated into 36 mL of G medium in 250 mL shake flasks and cultivated at 37 °C with shaking at 220 rpm. When OD600 reached 0.8–1.0 (in about 3 h), 0.1 mM IPTG was added. After 16 h of induction at 23 °C and 220 rpm, 4 mL of 20% glucose and certain volume of organic phase was added into the shake flasks. Dodecane (40 mL, 13.5 mL, and 4 mL) was added into cultures at dodecane ratios (v/v) of 1:1, 1:3, and 1:10. The organic solvents (40 mL) of olive oil and isopropyl myristate with a 1:1 ratio (v/v) was added into cultures. Cultivation was continued at 30 °C for 48 h or 72 h with shaking at 220 rpm.
The aqueous and organic phases were separated through centrifugation. Benzyl acetate, cinnamyl acetate and other metabolites in organic and/or aqueous phases were detected via HPLC. Data were collected from three independent experiments.
The overall product production of the two-phase cultures was calculated with respect to the aqueous phase volume (40 mL). The production of benzyl acetate or cinnamyl acetate in the organic phase was calculated by dividing total weight (mg or g) in organic phase sample by 40 mL. The total production of benzyl acetate or cinnamyl acetate was calculated by summing the production in the organic phase and in the aqueous phase.
Metabolite analysis
Metabolite concentrations were analyzed via HPLC (Agilent). A SilGreen C18 column (4.6 × 250 mm, 5 μm) (Greenherbs, Beijing, China) was utilized for HPLC analysis on a Shimadzu LC-20AD HPLC system, which was equipped with an SPD-M40 PDA detector. Solvent A was water with 0.1% formic acid, and solvent B was methanol. The flow rate of the mobile phase was 1 mL/min according to the following program: 0.01–2 min, 25% buffer B; 2–25 min, a linear gradient of buffer B from 25 to 100%; 25–35 min, 100% buffer B; 35–36 min, a linear gradient of buffer B from 100 to 25%; and 36–46 min, 25% buffer B. The sample injection volume was 20 µL. The signals of the metabolites were analyzed at 254 nm for BALC, BA, CA, cinnamyl alcohol, cinnamyl acetate, and benzyl acetate. BALC and cinnamyl acetate were identified via LC-HRMS.
The LC-HRMS analysis was carried out on an Agilent 1260 HPLC system coupled with an Agilent Infinity UV detector and a Bruker-MicrOTOF-II mass spectrometer that was equipped with an electrospray ionization device. Data acquisition and processing were done with MicrOTOF control version 3.0/Data Analysis Version 4.0 software. Optimized MS operating conditions were as follows: all spectra were obtained in positive mode over an m/z range of 50-1000 under a dry gas flow of 6.0 l min − 1, a dry temperature of 180 °C, a nebulizer pressure of 1 bar and a probe voltage of − 4.5 kV. The analysis programs were identical to HPLC analysis.
L-Phe was detected as its 2, 4-dinitrofluorobenzene derivative at 360 nm via HPLC. The HPLC program proceeded as follows: solvent A was water with 0.1% formic acid, and solvent B was acetonitrile; 0.01–5 min, 40% buffer B; 5–45 min, a linear gradient of buffer B from 50 to 100%; 45–55 min, 100% buffer B; 55–55.5 min, a linear gradient of buffer B from 100 to 40%; and 55.5–60 min, 40% buffer B. The sample injection volume was 20 µL.
Benzyl acetate in dodecane was identified via GC–MS analysis, which was performed on a Thermo Scientific Orbitrap Exploris GC 240 mass spectrometer (Thermo Fisher, Germany). A sample volume of 1 µL was injected into an S/SL injector, and compounds were separated using a TRACE 1310 gas chromatograph with TraceGOLD TG-5SILMS (30 m length × 0.25 mm inner diameter × 0.25 μm film thickness column). The oven temperature program was as follows: 70 °C for 2 min, followed by a ramp of 10 °C min− 1 to 325 °C, and an 8.5 min hold. The injection, transfer line, and ion source temperatures were 250 °C, 290 °C, and 250 °C, respectively. Accurate mass spectrometry data were acquired in a full-scan mode at 60,000 mass resolution (FWHM m/z 200) after a solvent delay of 3 min. Scan acquisition was performed to monitor all fragment ions within a mass range of 45–700 m/z. Raw peaks were extracted, peaks were aligned, deconvolution analysis was performed, peaks were identified, and the peak area was integrated using Thermo Scientific Xcalibur Version 4.5.445.18 software with the NIST2020 library. A mass window of ± 5 ppm was used to generate the extracted ion chromatograms (EICs); that is, only ions with a mass accuracy of ≤ 5 ppm was extracted.
Samples were diluted to fall within the standard curves if necessary. All experiments were conducted in triplicates and repeated at least twice. Titers were presented as means ± SD.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- CA:
-
Trans-Cinnamic Acid
- CA-CoA:
-
Cinnamoyl-CoA
- HPP-CoA:
-
3-Hydroxyphenylpropionyl-CoA
- KPP-CoA:
-
3-Ketophenylpropionyl-CoA
- BA-CoA:
-
Benzoyl-CoA
- BA:
-
Benzoic Acid
- BALD:
-
Benzaldehyde
- BALC:
-
Benzyl Alcohol
- CARs:
-
Carboxylic Acid Reductases
- ADHs:
-
Alcohol Dehydrogenases
- AKRs:
-
Aldo-Keto Reductases
References
Zainab Esmail Sadeq NSA-O, Omar G, Hammoodi AN, Abd. Anfal Salam Al-Mahdawi. Benzyl acetate: a review on synthetic methods. Eurasian J Phys Chem Maths. 2022;9:28–35.
Yadav GD, Mehta PH, Haldavanekar BV. Capsule membrane phase-transfer catalysis selective alkaline-hydrolysis and oxidation of benzyl-chloride to benzyl alcohol and benzaldehyde. Heterogen Catal Fine Chemicals Iii. 1993;78:503–12.
Pugh S, McKenna R, Halloum I, Nielsen DR. Engineering Escherichia coli for renewable benzyl alcohol production. Metab Eng Commun. 2015;2:39–45.
Otto M, Wynands B, Marienhagen J, Blank LM, Wierckx N. Benzoate synthesis from glucose or glycerol using engineered Pseudomonas taiwanensis. Biotechnol J. 2020;15:11.
Noda S, Kitazono E, Tanaka T, Ogino C, Kondo. A. Benzoic acid fermentation from starch and cellulose via a plant-like β-oxidation pathway in Streptomyces Maritimus. Microb Cell Fact. 2012;11:49.
Luo ZW, Lee SY. Metabolic engineering of Escherichia coli for the production of benzoic acid from glucose. Metab Eng. 2020;62:298–311.
Gahloth D, Aleku GA, Leys D. Carboxylic acid reductase. Structure and mechanism. J Biotechnol. 2020;307:107–13.
Huang XQ, Li R, Fu J, Dudareva N. A peroxisomal heterodimeric enzyme is involved in benzaldehyde synthesis in plants. Nat Commun. 2022;13:1352.
Gómez JL, Gómez MG, Murcia MD, Gómez E, Hidalgo AM, Montiel C, Martínez R. Biosynthesis of benzyl acetate: optimization of experimental conditions, kinetic modelling and application of alternative methods for parameters determination. Bioresource Technol Rep. 2020;11:100519.
Almeida SAAG, de Meneses AC, de Araújo PHH, de Oliveira D. A review on enzymatic synthesis of aromatic esters used as flavor ingredients for food, cosmetics and pharmaceuticals industries. Trends Food Sci Tech. 2017;69:95–105.
Seo H, Lee JW, Garcia S, Trinh CT. Single mutation at a highly conserved region of chloramphenicol acetyltransferase enables isobutyl acetate production directly from cellulose by at elevated temperatures. Biotechnol Biofuels. 2019;12:245.
Lee JW, Trinh CT. Microbial biosynthesis of lactate esters. Biotechnol Biofuels. 2019;12:1.
Choi KR, Luo ZW, Kim GB, Xu HW, Lee SY. A microbial process for the production of benzyl acetate. Nat Chem Eng. 2024;1:216–28.
Zhang MQ, Liu C, Xi DY, Bi HP, Cui ZZ, Zhuang YB, et al. Metabolic engineering of Escherichia coli for high-level production of salicin. Acs Omega. 2022;7:33147–55.
Xu D, Zhang L. Increasing agmatine production in Escherichia coli through metabolic engineering. J Agric Food Chem. 2019;67:7908–15.
Winkler M, Ling JG. Biocatalytic carboxylate reduction - recent advances and new enzymes. ChemCatChem. 2022;14:19.
Ling JG, Mansor MH, Abdul Murad AM, Mohd Khalid R, Quay DHX, Winkler M, et al. A functionally-distinct carboxylic acid reductase PcCAR4 unearthed from a repertoire of type IV CARs in the white-rot fungus pycnoporus cinnabarinus. J Biotechnol. 2020;307:55–62.
Brandenburger E, Braga D, Kombrink A, Lackner G, Gressler J, Künzler M, et al. Multi-genome analysis identifies functional and phylogenetic diversity of basidiomycete adenylate-forming reductases. Fungal Genet Biol. 2018;112:55–63.
Nancolas B, Bull ID, Stenner R, Dufour V, Curnow P. Atf1p is an alcohol acetyltransferase and a thioesterase. Yeast. 2017;34:239–51.
Seo H, Lee JW, Giannone RJ, Dunlap NJ, Trinh CT. Engineering promiscuity of chloramphenicol acetyltransferase for microbial designer ester biosynthesis. Metab Eng. 2021;66:179–90.
Liu GF, Huang L, Lian JZ. Alcohol acyltransferases for the biosynthesis of esters. Biotechnol Biofuels Bioprod. 2023;16:1.
D’Auria JC, Chen F, Pichersky E. Characterization of an acyltransferase capable of synthesizing benzylbenzoate and other volatile esters in flowers and damaged leaves of Clarkia breweri. Plant Physiol. 2002;130:466–76.
Van Moerkercke A, Schauvinhold I, Pichersky E, Haring MA, Schuurink RC. A plant thiolase involved in benzoic acid biosynthesis and volatile benzenoid production. Plant J. 2009;60:292–302.
Lee WH, Kim JW, Park EH, Han NS, Kim MD, Seo JH. Effects of NADH kinase on NADPH-dependent biotransformation processes in Escherichia coli. Appl Microbiol Biot. 2013;97:1561–9.
Qin N, Li L, Ji X, Li X, Zhang Y, Larsson C, Chen Y, Nielsen J, Liu Z. Rewiring central carbon metabolism ensures increased provision of acetyl-CoA and NADPH required for 3-OH-propionic acid production. ACS Synth Biol. 2020;9:3236–44.
Wang Y, Zhou S, Li R, Liu Q, Shao X, Zhu L, Kang MK, Wei G, Kim SW, Wang C. Reassessing acetyl-CoA supply and NADPH availability for mevalonate biosynthesis from glycerol in Escherichia coli. Biotechnol Bioeng 2022; 0.1002/bit.28,167.
Kruis AJ, Bohnenkamp AC, Patinios C, van Nuland YM, Levisson M, Mars AE, et al. Microbial production of short and medium chain esters: Enzymes, pathways, and applications. Biotechnol Adv. 2019; 37:7.
Sun L, Kwak S, Jin YS. Vitamin A production by engineered from xylose two-phase extraction. ACS Synth Biol. 2019; 8:2131–2140.
Lazzerini C, Domenici V. Pigments in extra-virgin olive oils produced in Tuscany (Italy) in different Years. Foods. 2017; 6:4.
Bruce LJ, Daugulis AJ. Solvent selection-strategies for extractive biocatalysis. Biotechnol Progr. 1991; 7:116–124.
Kaneko M, Ohnishi Y, Horinouchi S. Cinnamate: coenzyme A ligase from the filamentous bacterium Streptomyces coelicolor A3(2). J Bacteriol. 2003; 185:20–27.
Zhu L, Cui W, Fang Y, Liu Y, Gao X, Zhou Z. Cloning, expression and characterization of phenylalanine ammonia-lyase from Rhodotorula glutinis. Biotechnol Lett. 2013; 35:751–756.
Kallscheuer N, Vogt M, Kappelmann J, Krumbach K, Noack S, Bott M, et al. Identification of the phd gene cluster responsible for phenylpropanoid utilization in Corynebacterium glutamicum. Appl Microbiol Biot. 2016; 100:1871–1881.
Baez-Viveros JL, Osuna J, Hernandez-Chavez G, Soberon X, Bolivar F, Gosset G. Metabolic engineering and protein directed evolution increase the yield of L-phenylalanine synthesized from glucose in Escherichia coli. Biotechnol Bioeng. 2004; 87:516–524.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFC2105102), the National Natural Science Foundation of China (31770104), the Innovation fund of Haihe Laboratory of Synthetic Biology (NO. 22HHSWSS00023) and Competitive support of Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences (JZ1-ZG2-2301-025). We thank the senior engineer Yonghong Yao from the systems biology center of Tianjin institute of industrial biotechnology for technical help with GC-MS experiments and analyses.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
T. L conceived the project. T. Land H.Y supervised the project. H.Y and Q.K designed the experiments. Q. K, C. L, YJ. X and ZZ. C performed the experiments and collected the data. H.Y, Q. K, T. L and YB. Z wrote the manuscript. All authors have given approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Agreed by all authors.
Competing interests
This work has been included in a patent application by the Tianjin Institute of Industrial Biotechnology.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12934_2024_2513_MOESM1_ESM.docx
Supplementary Material 1: Supporting information accompanies this paper in Supplementary file. Fig. S1 L-Phe production and cell growth of strains U7A, U7AG and GAP. Fig. S2 HPLC analysis of the metabolites of strain EcBC04 harboring SrCAR. Fig. S3 Selection of optimal AATs for benzyl acetate biosynthesis. Fig. S4 HPLC analysis of the metabolites in organic phase when EcYZ07 was cultured using isopropyl myristate as in situ extractive agent in two-phase fermentation. Table S1. The primers used in construction of plasmids. Table S2 The sequence of genes with E. coli codon optimization in this study. Table S3 The conversion ratio of CARs towards BA and CA. Table S4 The production of benzyl acetate produced by the EcYZ01, EcYZ02 and EcYZ03 strains with 1:10 (v/v) ratio of dodecane overlay in two-phase in situ extractive fermentation. Table S5 The cell growth and benzyl acetate production of engineered EcYZ07 strain with the ratios (v/v) of dodecane overlay in two-phase in situ extractive fermentation. Table S6 The cell growth and benzyl acetate production of engineered EcYZ07 strain with 1:1 (v/v) ratio of olive oil and isopropyl myristate overlay in two-phase in situ extractive fermentation.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ke, Q., Liu, C., Zhuang, Y. et al. Metabolic engineering of Escherichia coli for high-level production of benzyl acetate from glucose. Microb Cell Fact 23, 238 (2024). https://doi.org/10.1186/s12934-024-02513-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-024-02513-y