
Abstract
The widespread use of molecular techniques in studying microbial communities has greatly enhanced our understanding of microbial diversity and function in the natural environment and contributed to an explosion of novel commercially viable enzymes. One of the most promising environments for detecting novel processes, enzymes, and microbial diversity is hot springs. We examined potential biases introduced by DNA preservation and extraction methods by comparing the quality, quantity, and diversity of environmental DNA samples preserved and extracted by commonly used methods. We included samples from sites representing the spectrum of environmental conditions that are found in Yellowstone National Park thermal features. Samples preserved in a non-toxic sucrose lysis buffer (SLB), along with a variation of a standard DNA extraction method using CTAB resulted in higher quality and quantity DNA than the other preservation and extraction methods tested here. Richness determined using DGGE revealed that there was some variation within replicates of a sample, but no statistical difference among the methods. However, the sucrose lysis buffer preserved samples extracted by the CTAB method were 15–43% more diverse than the other treatments.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The impact of molecular studies on our knowledge of microbial diversity cannot be overstated. As a consequence, the entire field of environmental microbiology, from basic ecological research into the organization of microbial communities to bioprospecting for commercially relevant enzymes has changed. Even with recent advances in culturing efforts [19, 38], the majority of microbes in the environment still cannot be cultivated in the laboratory [34]. However, inability to maintain an organism in culture is no longer a major impediment to accessing its genetic diversity. Metagenomic studies similar to those that have been useful in exploring the diversity of uncultivated organisms have also been used to mine for enzymatic diversity [22]. The biotechnology applications that are currently targeting microbial metagenomic studies range from the search for new antibiotics to environmentally sound biocatalysts such as amylases [26, 35].
Thermal environments have been a particularly rich source of unique organisms [2, 13, 16, 20, 43], processes [5], novel enzymes [19, 39], and on-going research into the origin and diversity of microbes. Fundamental to any of these studies is maximizing the detectable diversity by optimizing the quality and quantity of DNA examined and minimizing the biases of the methods. There are layers of potential bias in molecular studies because of the sequential nature of the process, including those inherent in PCR [44] which have been well studied and will not be addressed in this paper. The first potential source of bias in the molecular study of environmental samples is determined by the method used to preserve the biomass. Cultivation based studies have shown that the groups of organisms that can be cultured from samples change drastically if the sample is not adequately preserved [15]. However, the impact of sample preservation on molecular diversity surveys is rarely examined in the literature (except see [17]).
Another possible source of bias includes DNA extraction methods and post-extraction purification. The effect of DNA extraction method on detectable diversity has been examined in soil [10, 11], marine sediments [27], compost [23, 45], and volcanic environments [18]. The results of these studies are mixed; certain methods are more effective for a particular type of sample than others. Additionally, no reports have examined the impact that the range of environmental pH extremes may have on extraction efficiency, which is of particular importance for thermal samples since they can come from springs on either end of the pH spectrum. While there are plenty of studies that examine effectiveness of extraction method on a particular type of sample, we aim to determine the method that is best for a wide variety of sample types.
Here, we evaluated sample preservation and DNA extraction methods, to identify a procedure that results in high molecular weight DNA that is relatively free from contaminants and maximizes detectable diversity. We compared three preservation and four nucleic acid extraction methods. The ideal preservation and extraction method would work well with a variety of samples that included high and low biomass, a wide range of pH, as well as different sample types including microbial mats, filamentous biofilms, and sediments. Additionally, this method should be quick to accommodate high throughput of large sample numbers and facilitate sample collection from the remote backcountry where liquid nitrogen and dry ice are not practical. We included samples from the whole spectrum of environmental conditions that are found in Yellowstone National Park, USA (YNP) thermal features and our results are applicable to future studies in YNP, other extreme environments, and microbial surveys in general.
Methods and materials
Site description and sampling
Samples were collected from thermal features throughout YNP during the summer of 2002. A subset of fifteen samples was selected for this study that encompassed the full range of pH, temperature, and biomass types found in the park (YNP sample n = 15, see Table 1). The pH and temperature of the samples were split into three ranges, low (pH 0–4, temperature 40–60 °C), mid (pH 4.01–8, temperature 60.1–80 °C), and high (pH above 8.01, temperature above 80.1 °C). The samples were also categorized by the type of biomass collected: microbial mat, filaments, or sediment. Microbial mat samples are expected to be the highest biomass, for example, mats may contain up to 6.1 × 108 cells cm−3 and are also high in pigments and extracellular proteins and polysaccharides [4]. Filament samples represent an intermediate amount of biomass (maximum cell density in culture 4.6 × 107 cells ml−1 [33]) and are generally non-pigmented. The sediment samples are expected to be the lowest biomass (2.1–3.6 × 106 cells cm−3 [29]) and the highest clay content and heavy metal concentration [1]. Additionally, we collected samples from two neutral thermal springs in the Jemez Mountains, New Mexico. Samples (1–2.5 ml) were collected at each site with either sterile forceps or a syringe.
Sample preservation
Two replicate YNP samples were collected from each sampling site: one was preserved in an equal volume of sucrose lysis buffer (SLB) (20 mM EDTA, 200 mM NaCl, 0.75 M sucrose, 50 mM Tris-HCl, pH 9.0) [14] and the other replicate was preserved in an equal volume of GIT (5 M guanidine isothiocyanate, 50 mM Tris pH 7.4, 25 mM EDTA pH 8, 0.8% 2-mercaptoethanol)[6]. The YNP samples were held at ambient air temperature (10–26 °C) for up to 5 days before they were stored at −80 °C.
The two Jemez Springs, New Mexico samples were collected from neutral thermal springs located at the Jemez Springs Bath House (BH) and Giggling Star Resort (GS). Three replicate samples were collected. Two of the replicates were preserved in SLB, one of these replicates was frozen immediately in liquid nitrogen and the other was held at 20 °C for 7 days before being frozen at −80 °C. The third replicate sample was collected and mixed with molten 2% agarose while in the field for extraction the noodle method (see below).
Extraction methods
We used four extraction methods: lysis by pulse boil, a CTAB (hexadecyltrimethylammonium) extraction, high molecular weight DNA agarose noodle [42], and the Mo Bio Soil DNA Purification kit, which combined with the preservation methods results in six treatments. With the pulse boil method, nucleic acids were extracted from 200 μl of the YNP and Jemez samples that were preserved in SLB (referred to here as “Boil”) [36]. Briefly, the samples were boiled at 96 °C then cooled to 4 °C three times in a thermocycler, sodium dodecyl sulphate (SDS) (to a final concentration of 2%) and proteinase K (final concentration 250 μg ml−1) were added and the sample was incubated at 42 °C for 2.5 h, then incubated at 60 °C for 30 min, extracted once with phenol/chloroform then twice with chloroform, finally the DNA was precipitated and washed with ethanol.
We extracted nucleic acids from 200 μl of the YNP and Jemez SLB preserved samples using a variation of the CTAB method (referred to as CTAB S) [46]. Briefly, 2 volumes of 1% CTAB buffer (1% CTAB, 0.75 M NaCl, 50 mM Tris pH 8, 10 mM EDTA) and proteinase K (final concentration 100 μg ml−1) were added to the SLB preserved samples; incubated for one hour at 60 °C, SDS (final concentration 2%) was added and incubated one hour at 60 °C, extracted once with phenol/chloroform then twice with chloroform, finally the DNA was precipitated with ethanol. The YNP samples that were preserved in GIT (referred to as CTAB G) were also extracted using the CTAB method with the modification of washing the sample three times with filter sterilized water before the addition of CTAB to remove the GIT from the sample. GIT is a protein denaturant and inhibits proteinase K activity if not removed.
SLB (Mo Bio S) and GIT (Mo Bio G) preserved YNP samples were extracted using the Mo Bio Soil DNA purification kit following manufacturer’s suggested protocol (Mo Bio Laboratories, Inc., Solana Beach, CA).
Nucleic acids were extracted from YNP SLB preserved samples and all Jemez samples using the high molecular weight noodle extraction method (noodle) [42]. Briefly, the sample was mixed with molten 2% agarose and cooled in 1 mL syringes forming agarose noodles, the noodles were incubated for 3 h at 37 °C in a lysis buffer (10 mM Tris, 50 mM NaCl, 50 mM EDTA, 0.2% SDS, 1% Sarkosyl, 1 mg ml−1 lysozyme) then incubated at 37 °C in ESP buffer (1% Sarkosyl, 0.1 M EDTA, 1 mg ml−1 proteinase K), the ESP buffer was changed once a day for a total of 4 days, the noodles were then stored at 4 °C in TE storage buffer (10 mM Tris and 50 mM EDTA). The purified nucleic acids were extracted from the agarose noodles by incubating at 60 °C to melt the agarose then adding agarase and incubating overnight at 37 °C. The nucleic acids were purified from the agarose/agarase slurry by extracting once with phenol/chloroform then twice with chloroform. The nucleic acids were precipitated with ethanol.
Measuring extraction success
Environmental DNAs were electrophoresed on an ethidium bromide stained 1.2% agarose gel. The size and quality (evidence of shearing, etc.) of the DNA was evaluated using a size standard DNA ladder (EZ Load 1 Kb, BioRad Laboratories).
The purity of the extracted DNA was quantified by calculating the ratio of the absorbance at 260 and 280 nm (A260/A280). Nucleic acids extracted from environmental samples are often contaminated with humic organic carbons, metals, and other compounds which cause the DNA concentration calculated from 260 nm absorbance to be unreliable. To circumvent this problem, we also quantified the DNA using PicoGreen dsDNA Quantitation Reagent (Molecular Probes, Inc., Eugene, OR, USA). Extracted DNA (5 μl) was mixed with a 1:200 solution of PicoGreen in 1X tris-acetate buffer (TAE) and absorbance was read in a fluorometer.
The third parameter that was qualified was the ability to amplify 16S rRNA gene from the environmental genome of each extraction replicate. Dilutions of the genomic DNA were used as template. The reaction included (1X Promega buffer with 15 mM MgCl2, bovine serum albumin (0.04% final), 2.5 U Taq DNA polymerase (Promega U.S.) 2.5% Igepal CA-630 (Sigma–Aldrich), 10 μM each dATP, dGTP, dCTP, dTTP (BioLine USA, Inc.), 20 μM Bacterial specific primers [24] 338FGC (CGCCCGCCGCGCCCCGCGCCCGTCCCGCCGCCCCCGCCCTCCTACGGGAGGCAGCAG) and 519R (ATTACCGCGGCTGCTGG)). The PCR reaction (50 μl) was incubated in a thermocycler (ABI GeneAmp 2700) for 5 min at 94.0 °C then for 30 cycles of 30 s at 94.0 °C, 30 s at 50.0 °C and 30 s at 72.0 °C. The reaction was incubated at 72.0 °C for 7 min for final extension.
Richness
Differences in relative species richness among the preservation and extraction methods were determined using denaturing gradient gel electrophoresis (DGGE). Ten microlitre of PCR product (approximately 500 ng) was run on a 6% (wt/vol) acrylamide gel with 1X TAE (40 mM Tris, 20 mM acetate, and 1 mM EDTA) with a denaturing gradient of 20% (8% (vol/vol) formamide and 8.4% (wt/vol) urea) to 60% (24% formamide and 25.2% urea). The gels were run in a BioRad DCode Universal Mutation Detection System (BioRad Laboratories, Hercules, CA) at 180 V for 3.5 h. The gels were stained in 1X TAE containing SybrGreen (100 μl L−1) for 30 min then destained in 1X TAE for 15 min. The gels were photographed under UV light and DGGE bands were identified and analyzed using Kodak 1D software. For our analyses, only bands with a minimum intensity of 72% were recognized (program default). Bands were distinguished based on migration distance within each gel, as determined by the software using the DGGE product from Escherichia coli as a standard [9]. Although re-amplification and sequencing of individual DGGE bands can be particularly important in resolving heteroduplex fragments, we did not sequence the bands we detected in this study because we were interested in detecting the greatest number of bands possible. The effectiveness of methods within each sample was calculated by dividing the number of bands detected within a treatment by the total number of unique bands found within a sample. This calculation of relative species richness was computed for samples where at least two of the treatments resulted in successful amplification (YNP samples n = 12).
Statistical analysis
Statistical analyses were performed using Minitab software version 13 and SPSS 11 (for Mac OS X). The purity of the DNA among treatments was compared using the absolute value that resulted from subtracting the A260/A280 from 1.8, the A260/A280 of pure DNA [37]. Univariate ANOVA was used to detect statistical differences in the quantity and purity of the DNA and relative species richness (dependent variables) among the treatments and by pH, temperature, and biomass type (fixed factors). For analysis of the DGGE bands, only samples where at least two of the treatments amplified were included, otherwise a statistical comparison would not be possible. The Bonferroni test (which is more powerful for small sample sizes than Tukey’s test) was performed to identify treatments that were significantly different. Rarefaction curves were calculated in EstimateS version 8.0.0.
Results
Sample preservation
We extracted more DNA from the samples preserved in SLB than from the GIT replicates (paired t test p = 0.01). The extracted DNAs were quantified using the PicoGreen reagent. Using our protocol, the PicoGreen assay has a dsDNA detection limit of 500 pg ml−1. We also compared the amount of DNA recovered from the Jemez samples that were frozen or mixed with agarose in the field with those that were held at ambient temperature; there was no statistical difference between the two replicates (paired t test α = 0.05). Even when samples cannot be frozen immediately, the DNA is adequately preserved in SLB.
There was no significant difference among the preservation methods in the richness we detected (paired t test α = 0.05). However, the 16S rRNA gene from the samples that were preserved in SLB were amplified by PCR more successfully; 80.5% of SLB preserved samples PCR amplified compared to just 50% of the GIT preserved samples.
DNA extraction
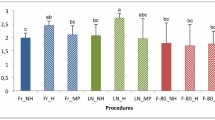
The CTAB extraction on SLB preserved samples resulted in the greatest yield of DNA. The results of the quantification are displayed in Fig. 1a. ANOVA showed a statistically significant difference in DNA yield among the extraction methods (p < 0.001). Post hoc multiple comparison analysis indicates that the CTAB S extracted samples resulted in the highest quantity of DNA.

a Mean DNA concentration as measured using PicoGreen assay for each preservation and extraction method with 95% confidence intervals, outliers are filled circles. b Mean DNA purity with 95% confidence interval and outliers. Pure DNA has a A260/A280 ratio of 1.8, shown as the gray bar. The concentrations of the DNAs from the noodle extractions were not high enough to be detected by the fluorometer
DNA purity, as measured by A260/A280 ratio, was greatest in samples extracted using the Mo Bio extraction kits, however, the kit results in less DNA and smaller fragments (presumably from shearing and the filter column used in the kit [3]) (Fig. 1b). There was no significant difference in the purity of the DNA extracted with the Boil, CTAB S, or CTAB G methods (ANOVA α = 0.05). The noodle method did not result in sufficient DNA quantity to obtain reliable absorbance data, therefore it was not included in the DNA purity comparison. Most of the amplification success we observed was in sample extracted with either the CTAB method (89% amplified) or the Mo Bio kit (95% amplified) (ANOVA and Bonferroni test p < 0.001).
The sequence diversity detected by DGGE, number of bands, varied within the replicates of the samples (Fig. 2). The only statistically significant difference we detected was between the CTAB S and the noodle method (p = 0.014). However, CTAB S resulted in 15–43% more bands than the other treatments (Fig. 3). There were no differences at the fixed effects level (pH, temperature, or biomass type), nor were any interactions (e.g., pH X method) among the factors detected. This suggests that CTAB S is appropriate for a wide range of sample types.

Example DGGE of PCR products on 6% acrylamide gel with 20–60% urea/formamide denaturing gradient, stained with SybrGreen [Treatments: a Boil, b CTAB S, c CTAB G, d Mo Bio S, e Mo Bio G, f noodle]. Samples marked with an asterisk were frozen in the field

a Rarefaction curves of DGGE bands detected with each extraction method. The 95% confidence interval for the diversity of all samples and extraction methods combined is in gray to demonstrate that there was no significant difference in diversity detected by each method. b Mean diversity detected as number of DGGE bands with 95% confidence intervals, outliers are filled circles
Discussion
Sample preservation
SLB preserved our extreme environment samples better than the GIT solution. This was likely due to more effective lysing, as the SLB initiates the lysis process while the sample is being stored. Additionally, the alkaline SLB raised the pH within the sample vials, potentially slowing the degradation of the DNA in low pH samples. Raising the pH of acidic samples is likely to have two positive impacts on preserving the DNA in those samples. First, DNA degrades in acidic solutions due to depurination [12], raising the pH of the solution reduces the rate of this reaction. Second, DNA binding to clay minerals increases as the pH decreases [21]. Even when samples could not be frozen immediately, the DNA is adequately preserved in SLB. This result is especially important because of restrictions on carrying and shipping dry ice and for samples that are collected from remote areas where it is logistically very difficult to freeze samples immediately and maintain them frozen. The non-toxic nature of SLB may be an additional attraction for field microbiologists, and for those shipping samples either commercially or hand carrying on airplanes. Although this study did not assess the relative effectiveness of the different methods in extracting RNA, we have successfully produced cDNA from samples preserved in SLB using reverse transcriptase PCR (unpublished data).
Previous studies have suggested that the noodle method, for very high molecular weight DNA [42], and storage in absolute ethanol, for samples where immediate freezing is not possible [17], adequately preserve DNA. However, based on the results from this study, the noodle preservation method does not work well with the samples we collect which often have low biomass in a dense extracellular matrix. In many samples, the concentration of DNA recovered using the noodle method was an order of magnitude less than recovered using the CTAB protocol. Similarly, we did not test ethanol preservation because the ethanol must be nearly absolute to prevent DNA degradation [8, 28] which would not be possible for samples that contain significant amounts of water. Therefore, we do not think that either the noodle method or absolute ethanol preservation are appropriate for thermal environmental samples.
DNA extraction efficiency and purity
When considering preservation alone, among the techniques we tested, SLB with or without freezing preserved environmental DNA best. However, we also were interested in the effect of extraction method on the amount and quality of extracted DNA and detectable diversity. Extracting pure DNA from environmental samples is nearly as important as extraction efficiency and is one of the most difficult technical problems in using molecular techniques on environmental samples. Most DNA extraction procedures co-extract humic organic carbon, pigments, heavy metals, and other contaminants. These contaminants play havoc with PCR reactions and can degrade the DNA during storage. We found that CTAB extraction combined with SLB preserved samples resulted in the most DNA. The greater efficiency of the CTAB extraction method has previously been found in several environments, marine sediments [27], basalt rock [18], and by one of us in caterpillar intestinal tracts and filtered water (unpublished data) indicating that this method is effective on a wide range of sample types. An extraction comparison on compost microbial communities found no difference in the amount of DNA recovered by the CTAB method, but it resulted in the highest percentage of cells lysed [45]. It is likely that the combination of proteinase K and hot SDS lyses more cells, including cell types more resistant to lysis (e.g., gram-positive bacteria) [32]. We found two exceptions where larger quantities of DNA were not recovered by the CTAB method. The PicoGreen analysis on two samples, 066-MV and 139-LS, indicates that more DNA was extracted using the Mo Bio kit than was extracted with the Boil and CTAB methods. The Boil and CTAB extracted DNA from these two samples was dark brown, which was most likely caused by pigments that were co-extracted with the DNA. The Mo Bio kit, which produced clear DNA extracts, was effective at removing these organics because it is the only method that incorporated a post-extraction purification [3]. Work on basalt rock samples found that only Mo Bio extracted DNA could be amplified [18], which is contrary to our results. We suspect that the co-extracted pigments interfered with PicoGreen fluorescence, but both of these samples were successfully amplified with all of the extraction methods. The co-extracted pigments from the Boil and CTAB methods do not appear to inhibit the PCR reaction. It should be noted that some of the samples with no detectable DNA, as determined using the PicoGreen fluorometer quantification, still had enough DNA to amplify with PCR (theoretical detection limit for PCR amplification <1 pg ml−1[41]).
Richness
Although the CTAB S treatments consistently had more bands than any of the other methods (15–43%), the only statistical difference detected was between the CTAB S and the noodle extraction method. We also tested for fixed effects such as pH, temperature, and biomass type, but did not detect any statistical differences. However, there were three samples that we were unable to amplify using two or more of the extraction treatments, those samples could not be included in the richness analyses. It is interesting to note that all three of these samples were sediments from low pH systems (<pH 3.5), indicating that there may be a sample type or geochemistry effect on extraction efficiency or amplification success. This result could have been due to inefficient extraction of DNA, co-extraction of PCR inhibitors, or too little target DNA in the original sample (i.e., if the community was dominated by Archaea to the exclusion of Bacteria). For those types of extremely recalcitrant samples, it may be necessary to crush the sample using a sterile mortar before the enzymatic lysis step [18]. However, CTAB worked on a majority of the samples and we suggest it should be used as a good general extraction method. If unsuccessful, alternate approaches should be used that may more effectively lyse the biomass or better remove humics.
There are many methods for assessing microbial community richness, generally based on the 16S rRNA gene, such as clone libraries, DGGE, and T-RFLP (terminal restriction fragment length polymorphisms). These analyses have greatly increased our understanding of microbial ecology and revealed novel enzymes. DGGE is an attractive method for microbial ecology studies because of the relatively rapid ease with which a community profile can be generated. DGGE can resolve minor base differences among samples; therefore, distinct bands may represent sequences that differ by less than 1% [31]. Depending on the nature of the study being performed, 1% may or may not be significant to the research question. Conversely, we are aware that individual bands could be heteroduplexes [7, 40], comprised of several different sequences, however, because our methods were standardized across samples, this effect does not figure in our conclusions. We repeated the PCR amplification and DGGE for selected samples to ensure reproducibility in the number of bands detected (data not shown, but available on request).
It is standard practice that investigators informally evaluate methods to determine which are appropriate for their study. However, few of these studies have been published [11] and often, statistical analyses are not used to determine the most appropriate methods [9, 18]. Additionally, other published studies did not evaluate the effect of extraction method on the diversity detected, only the quantity of DNA [25, 30]. This study presents a framework for the systematic comparison and evaluation of methods by statistical analysis. The current interest in the organisms that inhabit extreme environments makes this study, which used samples from a wide variety of thermal environments, of importance to thermal biology researchers, and applicable to other microbial studies in general.
References
Allen ET, Day AL (1935) Hot springs of the Yellowstone National Park. Carnegie Institution, Washington, DC
Barns SM, et al. (1994) Remarkable archaeal diversity in a Yellowstone National Park hot spring environment. Proc Natl Acad Sci USA 91(March):1609–1613 doi:10.1073/pnas.91.5.1609
Braid MD, et al. Testing the UltraClean soil DNA purification kit on a diverse range of soils by PCR amplification of 16S rDNA. In: ASM General Meeting. 1999
Brock TD Thermophilic microorganisms and life at high temperatures. 1978, Springer-Verlag, New York, pp 465
Bryant DA et al (2007) Candidatus Chloracidobacterium thermophilum: an aerobic phototrophic acidobacterium. Science 317:523–526. doi:10.1126/science.1143236
Cary SC et al (1993) Identification and localization of bacterial endosymbionts in hydrothermal vent taxa with symbiont-specific polymerase chain reaction amplification and in situ hybridization techniques. Mol Mar Biol Biotechnol 2(1):51–62
Ferris MJ, Ward DM (1997) Seasonal distributions of dominant 16S rRNA-defined populations in a hot spring microbial mat examined by denaturing gradient gel electrophoresis. Appl Environ Microbiol 63(4):1375–1381
Frantzen MAJ et al (1998) Empirical evaluation of preservation methods for faecal DNA. Mol Ecol 7:1423–1428. doi:10.1046/j.1365-294x.1998.00449.x
Fromin N et al (2002) Statistical analysis of denaturing gel electrophoresis (DGE) fingerprinting patterns. Environ Microbiol 4(11):634–643. doi:10.1046/j.1462-2920.2002.00358.x
Frostegard A et al (1999) Quantification of bias related to the extraction of DNA directly from soils. Appl Environ Microbiol 65(12):5409–5420
Gabor EM, de Vries EJ, Janssen DB (2003) Effecient recovery of environmental DNA for expression cloning by indirect extraction methods. FEMS Microbiol Ecol 44:153–163. doi:10.1016/S0168-6496(02)00462-2
Garrett RH, Grisham CM (1995) In: Field C (ed) Biochemistry, Saunders College Publishing, Philadelphia
Ghosh D et al (2003) Molecular phylogenetic exploration of bacterial diversity in a Bakreshwar (India) hot spring and culture of Shewanella-related thermophiles. Appl Environ Microbiol 69(7):4332–4336. doi:10.1128/AEM.69.7.4332-4336.2003
Giovannoni SJ et al (1990) Tangential flow filtration and preliminary phylogenetic analysis of marine picoplankton. Appl Environ Microbiol 56:2572–2575
Haldeman DL et al (1994) Changes in bacteria recoverable from subsurface volcanic rock samples during storage at 4°C. Appl Environ Microbiol 60(8):2697–2703
Harris KJ, Kelley ST, Pace NR (2004) New perspective on uncultured bacterial phylogenetic division OP11. Appl Environ Microbiol 70(2):845–849. doi:10.1128/AEM.70.2.845-849.2004
Harry M, Gambier B, Garnier-Sillam E (2000) Soil conservation for DNA preservation for bacterial molecular studies. Eur J Soil Sci 36:51–55
Herrera A, Cockell CS (2007) Exploring microbial diversity in volcanic environments: a review of methods in DNA extraction. J Microbiol Methods 70:1–12. doi:10.1016/j.mimet.2007.04.005
Hobel CFV et al (2004) Use of low nutrient enrichments to access novel amylase genes in silent diversity of thermophiles. World J Microbiol Biotechnol 20:801–809. doi:10.1007/s11274-004-2623-4
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180(18):4765–4774
Khanna M, Stotzky G (1992) Transformation of Bacillus subtilis by DNA bound on montmorillonite and effect of DNase on the transforming ability of bound DNA. Appl Environ Microbiol 58(6):1930–1939
Kowalchuk GA et al (2007) Finding the needles in the metagenome haystack. Microb Ecol 53:475–485. doi:10.1007/s00248-006-9201-2
LaMontagne MG et al (2002) Evaluation of extraction and purification methods for obtaining PCR-amplifiable DNA from compost for microbial community analysis. J Microbiol Methods 49:255–264. doi:10.1016/S0167-7012(01)00377-3
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, (eds) Nucleic acid techniques in bacterial systematics. Wiley, Chichester, pp 115–175
Leuko S et al (2008) Lysis efficiency of standard DNA extraction methods for Halococcus spp. in an organic rich environment. Extremophiles 12:301–308. doi:10.1007/s00792-007-0124-8
Lorenz P, Eck J (2005) Metagenomics and industrial applications. Nat Rev Microbiol 3:510–516. doi:10.1038/nrmicro1161
Luna GM, Dell’Anno A, Danovaro R (2006) DNA extraction procedure: a critical issue for bacterial diversity assessment in marine sediments. Environ Microbiol 8(2):308–320. doi:10.1111/j.1462-2920.2005.00896.x
Mandrioli M, Borsatti F, Mola L (2006) Factors affecting DNA preservation from museum-collected lepidopteran specimens. Entomol Exp Appl 120(3):239–244. doi:10.1111/j.1570-7458.2006.00451.x
Meyer-Dombard DR, Shock EL, Amend JP (2005) Archaeal and bacterial communities in geochemically diverse hot springs of Yellowstone National Park, USA. Geobiology 3:211–227. doi:10.1111/j.1472-4669.2005.00052.x
Miller DN et al (1999) Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl Environ Microbiol 65(11):4715–4724
Moeseneder MM et al (1999) Optimization of terminal-restriction fragment length polymorphism analysis for complex marine bacterioplankton communities and comparison with denaturing gradient gel electrophoresis. Appl Environ Microbiol 65(8):3518–3525
More MI et al (1994) Quantitative cell lysis of indigenous microorganisms and rapid extraction of microbial DNA from sediment. Appl Environ Microbiol 60(5):1572–1580
Nakagawa S et al (2005) Sulfurihydrogenibium yellowstonense sp. nov., an extremely thermophilic, facultatively heterotrophic, sulfur-oxidizing bacterium from Yellowstone National Park, and emended descriptions of the genus Sulfurihydrogenibium, Sulfurihydrogenibium subterraneum and Sulfurihydrogenibium azorense. Int J Syst Evol Microbiol 55:2263–2268. doi:10.1099/ijs.0.63708-0
Pace NR (1997) A molecular view of microbial diversity and the biosphere. Science 276:734–740. doi:10.1126/science.276.5313.734
Pontes DS et al (2007) Molecular approaches: advantages and artifacts in assessing bacterial diversity. J Ind Microbiol Biotechnol 34:463–473. doi:10.1007/s10295-007-0219-3
Reysenbach AL, Ehringer M, Hershberger K (2000) Microbial diversity at 83 degrees C in Calcite Springs, Yellowstone National Park: another environment where the Aquificales and “Korarchaeota” coexist. Extremophiles 4(1):61–67
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a Laboratory manual. 2nd edn, Plainview, Cold Spring Harbor Laboratory Press, New York
Schoenborn L et al (2004) Liquid serial dilution is inferior to solid media for isolation of cultures representative of the phylum-level diversity of soil bacteria. Appl Environ Microbiol 70(7):4363–4366. doi:10.1128/AEM.70.7.4363-4366.2004
Schoenfeld T, et al. (2008) Viral diversity and improved DNA polymerases, In: YNP RCN/TBI workshop, Mammoth Hot Springs, WY, USA
Speksnijder AGCL et al (2001) Microvariation artifacts introduced by pcr and cloning of closely related 16S rRNA gene sequences. Appl Environ Microbiol 67(1):469–472. doi:10.1128/AEM.67.1.469-472.2001
Steffan RJ, Atlas RM (1998) DNA amplification to enhance detection of genetically engineered bacteria in environmental samples. Appl Environ Microbiol 54(9):2185–2191
Stein JL et al (1996) Characterization of uncultivated prokaryotes: isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. J Bacteriol 178(3):591–599
Takacs-Vesbach CD et al (2008) Volcanic calderas delineate biogeographic provinces among Yellowstone thermophiles. Environ Microbiol 10(7):1681–1689. doi:10.1111/j.1462-2920.2008.01584.x
von Wintzingerode F, Gobel UB, Stackebrandt E (1997) Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21(3):213–229. doi:10.1111/j.1574-6976.1997.tb00351.x
Yang ZH et al (2007) Comparison of methods for total community DNA extraction and purification from compost. Appl Microbiol Biotechnol 74:918–925. doi:10.1007/s00253-006-0704-z
Zhou J, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microbiol 62(2):316–322
Acknowledgments
We are grateful for the comments and input by Robert Sinsabaugh and Diana Northup who kindly reviewed an earlier version of the manuscript. This work was supported by NSF Biodiversity Surveys and Inventories grant 02-06773 to CTV.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mitchell, K.R., Takacs-Vesbach, C.D. A comparison of methods for total community DNA preservation and extraction from various thermal environments. J Ind Microbiol Biotechnol 35, 1139–1147 (2008). https://doi.org/10.1007/s10295-008-0393-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-008-0393-y