
Abstract
Bacteria account for a major proportion of Earth’s biological diversity. They play essential roles in quite diverse environments and there has been an increasing interest in bacterial biodiversity. Research using novel and efficient tools to identify and characterize bacterial communities has been the key for elucidating biological activities with potential for industrial application. The current approach used for defining bacterial species is based on phenotypic and genomic properties. Traditional and novel DNA-based molecular methods are improving our knowledge of bacterial diversity in nature. Advances in molecular biology have been important for studies of diversity, considerably improving our knowledge of morphological, physiological, and ecological features of bacterial taxa. DNA–DNA hybridization, which has been used for many years, is still considered the golden standard for bacteria species identification. PCR-based methods investigating 16S rRNA gene sequences, and other approaches, such as the metagenome, have been used to study the physiology and diversity of bacteria and to identify novel genes with potential pharmaceutical and other biotechnological applications. We examined the advantages and limitations of molecular methods currently used to analyze bacterial diversity; these are mainly based on the 16S rRNA gene. These methods have allowed us to examine microorganisms that cannot be cultivated by routine methods and have also been useful for phylogenetic studies. We also considered the importance of improvements in microbe culture techniques and how we can combine different methods to allow a more appropriate assessment of bacterial diversity and to determine their real potential for industrial applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The biosphere was formed by and is completely dependent on the metabolism of microorganisms and on their interactions with each other. Currently, it is estimated that there are about 4–6 × 1030 different prokaryotic cells, exceeding, by various orders of magnitude, all plant and animal diversity [124]. Bacterial cells arose about 3.8 billion years ago; they are microscopic, morphologically simple and are widespread throughout all environments, including those with extreme conditions [3, 44, 55, 69, 97, 103, 114]. The long history and the importance of microorganisms help explain the great morphological, physiological, and genetic diversity of these forms of life [114]. This enormous genetic variability is the result of rare mutations and recombination events, which allow them to reply to environmental changes. Bacteria can exchange and acquire genes from distantly related organisms by horizontal gene transfer (HGT), consequently increasing rates of speciation, which can be considerably higher than in eukaryotes [14, 52, 119, 123].
The fundamental unit of biological diversity and the basis of taxonomic hierarchy is the species. However, most bacteria cannot be isolated and cultured with current techniques; consequently, natural bacterial diversity is still relatively unexplored. For more than 100 years, microorganisms have been described and identified by culture methodologies [36, 62, 97, 103]. During this period, there has been immense progress in clinical microbiology, much more than in environmental microbiology, due to the importance of microorganisms for public health. Recently, the astonishing diversity of microorganisms has been discovered, along with their importance in providing environmental services essential for sustained life on Earth. Bacteria play important roles in biogeochemical cycles (carbon, nitrogen, and other minerals), bioremediation processes, energy conversion, biocatalysis, and natural product synthesis; this makes bacteria important potential resources for new industrial and biomedical processes [9, 71, 24]. Consequently, there has been increasing interest in the exploration of natural bacterial communities and their beneficial aspects; such studies are required to fulfill the promise of bacterial biotechnology.
Bacterial species are regarded as a genomically coherent group sharing a high degree of similarity in several different phenotypic and genomic properties that have been characterized through a polyphasic approach. However, knowledge about natural bacterial diversity is still limited, and there are only 6,373 validly described species [32] (for updates see http://www.bacterio.cict.fr). The difficulties and limitations of the methods available for culturing bacteria found in natural environments, such as soil and freshwater, has made the study of microbial diversity a difficult and complex effort [36, 64, 84, 97, 98, 114]. Nevertheless, during the last few years, advances and improvement of molecular techniques based on DNA sequencing and analysis of ribosomal gene sequences (mainly of small subunit 16S rRNA- SSU rRNA) of various prokaryotes has provided data that give considerable information on taxonomic relationships, ecological roles and the evolution of bacterial species found in environmental samples, without the need for isolation and culture. These new techniques have uncovered new bacterial functions and have revolutionized our concept of the value of microbial diversity, which till now has been largely undescribed [20, 27, 35, 36, 59, 63, 64, 72, 83, 97, 98, 114, 125]. Hence, bacterial ecology and industrial microbiology have come together; biodiversity studies have potential for important discoveries in both fields.
Approaches used to identify bacterial species
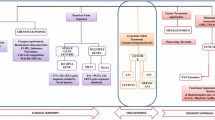
At present, it is widely accepted that it is necessary to use various methods to identify and characterize bacteria species and to study and determine the diversity of this domain. A combination of many different methodologies has been directed toward analyzing phenotypic, genomic, and phylogenetic characteristics for taxonomic purposes; this combination is defined as a ‘polyphasic approach’ (Fig. 1) [5, 15, 20, 37, 98, 117]. In practice, the meaning of specific tests has been influenced by how they correlate with DNA analysis for the identification of a particular bacterium.

Useful culture dependent and culture independent methods. RFLP restriction fragment length polymorphism, ARDRA amplified rDNA restriction analysis, DGGE denaturing gradient gel electrophoresis, TGGE temperature gradient gel electrophoresis, SSCP single-stranded conformation polymorphism, FISH fluorescence in situ hybridization
Characterizing phenotypes
The classical phenotypic tests have been used for morphological, physiological, and biochemical identification of bacteria and for biotechnological applications [44, 109]. Substrate utilization and bacterial product production profiles provide important data for studies of functional biodiversity. Phenotypic characterization has proven useful to study and characterize genes that encode proteins, which could have industrial applications (Table 1).
A combination of phenotypic and genomic information is required for correct description and classification of bacteria [15, 99, 113]. Recently, significant developments in various areas (chemistry, molecular biology, and bioinformatics) have provided new techniques of improving our knowledge of microbial diversity, as well as facilitating taxonomic and biotechnological studies [82, 99].
DNA–DNA hybridization
Whole genome DNA–DNA hybridization has been used for bacterial genotypic characterization for decades, increasing the quality and quantity of information useful for identification, which was previously limited to phenotypic properties [99]. DNA–DNA hybridization was one of the first molecular researches [18] and until now remains a cornerstone for bacterial species delineation [108]. This standard technique allows for comparison and measurement of genomic similarities between the total genome of two species under standardized conditions. A group of strains showing 70% or greater DNA–DNA similarities and with 5°C or less in thermal stability between homologuous and heterologuous heteroduplexes is considered to be the same species [45, 58, 117, 122]. Nevertheless, this method is arbitrary in its ability to evaluate the actual sequence similarity between two whole genomes and to suitably describe bacterial species [87, 90, 117]. Additionally, DNA–DNA hybridization studies are time-consuming; they allow the study of only a few bacterial clusters and are not applicable to uncultured organisms, which account for the majority of living prokaryotes [37, 58, 80, 87]. Despite the drawbacks and limitations of this method, it has been one of the most important approaches in bacterial species circumscription, as shown by Mechichi et al. [79, 108]. They described two new species of the genus Thauera and one species of the genus Azoarcus, using a polyphasic approach. They used DNA–DNA hybridization, which is an essential step for the differentiation of strains that have been isolated from different environments. These strains degrade aromatic compounds, such as phenol, benzoate, and toluene. The characterization of these three new species demonstrates the importance of genome research and it will be extremely important for analysis of novel aromatic catabolic functions and for the use of these bacteria as biocatalysts for the biodegradation and biotransformation of aromatic compounds [79].
Considering the significant role of DNA–DNA hybridization assay and the difficulty to implement this technique in routine laboratories, novel and alternative methods, such as melting profiles in microplates, random genome fragments, and DNA microarrays have been proposed to improve or supplement this approach [11, 80, 85]. Microarrays have potential as a rapid technique for environmental and phylogenetic studies, allowing the analysis of a large number of samples. An example of application is the rapid detection of 90 antibiotic resistance genes in 36 strains of Gram-positive bacteria (Bacillus, Clostridium, Enterococcus, Lactococcus, Lactobacillus, Listeria, Staphylococcus, and Streptococcus). Using this technique, three different genes involved in erythromycin resistance were detected in Staphylococcus haemolyticus and three genes carried by transposon Tn5405 were detected in Clostridium perfringens. These results demonstrated that microarrays make rapid screening of antibiotic resistance genes possible and that they can be useful for industrial and biomedical applications [93].
RNA ribosomal (rRNA) genes
Ribosomal RNAs are ancient molecules, being primordial participants of cell protein production machinery; they have a ubiquitous distribution, are conserved between organisms that are phylogenetically distant and are not affected by environmental changes [99]. Ribosomal RNA genes can occur in variable numbers in different organisms and are almost unaffected by horizontal genetic transference mechanisms. The combination of these properties makes these genes suitable for studies of microbial evolution and phylogeny [1, 12].
Bacteria have three genes that code 5S, 16S, and 23S rRNAs, essential components of ribosomes involved in the translation of messenger RNA (mRNA) for protein synthesis. These genes are typically organized into an operon that contains internal transcribed spacers (ITS), which vary widely in length and sequences [1, 34, 61, 65]. Spacer regions, especially those located between the 16S and 23S rRNA genes, have more genetic variation than the other regions that code rRNA genes, due to variation in length and in the number of tRNA genes contained in the sequences. ITS polymorphism can be useful for differentiation of closely related bacterial genera and species [13, 20, 25, 61]. Bacterial species can have up to 15 copies of the ribosomal operon in their genome [1, 34, 65].
Analysis of bacterial diversity
The 16S rRNA gene has several characteristics that favor its use for molecular studies, including suitable length, about 1,500 bp, the highly conserved regions among different and distant species and ease of its manipulation [4, 99]. This gene is generally weakly affected by HGT; currently, there is considerable information available in databases due to its extensive and growing utilization in microbial taxonomic studies. The properties of the 16S rRNA gene make it a suitable tool for phylogenetic inferences [99].
Woese in 1970 proposed a phylogenetic classification system for prokaryotic species based on the divergence of small subunit ribosomal RNA sequences (16S rRNA for prokaryotes and 18S rRNA for eukaryotes). Comparative analysis of SSU rRNA can elucidate evolutionary relationships and diversity among organisms. Woese introduced a view of the tree of life divided into three domains: Bacteria and Archaea, both classified as prokaryotes, and Eukarya. Inside the Bacteria domain, he described 11 divisions based on nucleotide sequences of 16S rRNA of cultured microorganisms [125]. This phylogenetic system, based on analysis of 16S rRNA gene sequences, opened new perspectives and became a useful model for studies of evolution and relationships among the various existing species [77]. There has been interest in examining the enormous unknown diversity of prokaryotes present in different ecosystems, based on information about the 16S rRNA gene in microorganisms that cannot be isolated in culture. Finding <97% 16S rRNA gene sequence similarity in comparison with known species has been regarded as a criterion for a new species description.
Today, it is believed that more than 53 divisions exist in the Bacteria domain, identified mainly through phylogenetic analysis of environmental samples that cannot be cultured, based on 16S rRNA gene sequences. However, most of the bacteria described in these new divisions based on molecular analysis are not cultivable [63]. This opens possibilities for obtaining evolutionary and phylogenetic information about new microorganisms, but frequently gives little information about their functional role, ecological relevance and genetic information about the different species of the community [54, 63, 81, 97, 104, 111].
Polymerase chain reaction (PCR)
PCR technology was developed in 1985 by Kary Mullis, and has had such a strong impact and has been so incredibly useful in many areas of science that the inventor received a Nobel Prize. Amplification of DNA by PCR has become extremely useful for bacterial detection in heterogeneous samples. Exploring bacterial biodiversity by PCR is the most commonly used method in studies of 16S rRNA genes; the information that is obtained can be compared with that from other techniques through pattern analysis (Table 2), including ARDRA, amplified rDNA restriction analysis [38, 68, 126]; DGGE, denaturant-gradient gel electrophoresis [106]; TGGE, temperature-gradient gel electrophoresis [8]; RFLP, restriction fragment length polymorphism [73]; SSCP, single-stranded conformation polymorphism [40]; evaluation of sample size and replication; FISH, fluorescence in situ hybridization [2]; cloning and sequencing; probe hybridization and microarrays. These techniques allow rapid and sensitive detection of bacteria diversity independent of culture methods and can also be used to identify metabolically active compounds if the desired gene is conserved enough to allow the design of specific primers.
PCR-based DNA fingerprinting provides information about differences in bacterial populations. Over the last decade, there has been increasing interest in studying sulfate-reducing bacteria (SRB), which play an important role in the degradation of organic matter, an essential process in ecosystem and environmental remediation. Nested-PCR-DGGE was used to study the diversity of SRB in samples from mixed microbial communities, with high resolution and reproducibility. The knowledge acquired in this study proved to be useful in bioremediation processes and for studies to improve pollutant removal efficiency [21].
Aerobic thermophilic bacteria diversity isolated from Irish soils was analyzed by ARDRA; sequencing of 16S rRNA genes showed that an abundance of genetically different members of the genus Geobacillus predominated in the sample. Geobacillus species were found to be sources of diverse compounds (proteases, lipases, amylases, and pullanases) with biotechnological applications and of interest for industry. McMullan et al. described the capacity of Geobacillus isolates to metabolize herbicides and showed that these isolates are potential sources of genes that would be useful for agricultural biotechnology [78].
Sequencing of the 16S rRNA gene and comparative analysis of the sequences is useful for understanding phylogenetic relationships among prokaryotes above the species level. Nevertheless, as the more information we have about 16S rRNA gene sequences, the more evident it becomes that they have limitations in their use in studies to distinguish closely related but ecologically distinct bacteria [90, 92]. Approaches based only on 16S rRNA gene sequences are insufficient to classify prokaryotic species and do not reveal the true relationships among the genomes of microorganisms.
Limitations of 16S rRNA sequence analysis
The finding of more than one 16S rRNA gene sequence in a single genome and variation in operons among strains of the same species provides critical information on bacterial diversity and evolution. The heterogeneity of 16S rRNA gene sequences among operons of the same genome is not an indication of the actual bacterial diversity [1, 19, 37, 105, 121]. Thermophilic microorganisms have the highest divergence among 16S rRNA gene sequences within a single genome; analyses have suggested that the rrnC operon was acquired by HGT. Generally, the divergence among 16S rRNA gene sequences in the same genome is less than 1% [1]. High similarity can be found in 16S rRNA gene sequences among some closely related microorganisms that lack resolution at the species level, as has been described for Bacillus, Ochrobactrum, Enterobacter and Taylorella [49, 67, 75, 95]. The considerable stability of 16S rRNA gene sequences and the low rate of evolution of this gene sometimes do not permit the identification of ecotypes; consequently, other techniques that examine genomic and physiological diversities and ecological niches may need to be applied [60, 70, 116]. Jaspers and Overmann [60] isolated 11 strains of Brevundiomas alba with identical 16S rRNA gene sequences. Using a combination of methods, they found great genetic diversity among strains from distinct populations with genetically determined adaptations and concluded that these strains probably occupy different ecological niches. Another limitation is the use of a universal pair of primers for amplification of 16S rRNA gene sequences in studies of bacterial diversity. PCR artifacts, such as preferential amplification of certain sequence types, generation of chimeric sequences and false positives due to experimental contaminants, can give inaccurate information about the actual diversity of microbial communities in environmental samples [4, 20, 28, 34, 51].
New approaches to access bacterial diversity
Approaches based on housekeeping genes that exist in a single copy have been suggested due to the heterogeneity of 16S rDNA and the implications of this heterogeneity for studies of bacterial diversity and phylogeny [20, 37]. Hence, the use of other genes, such as rpoB, gyrB, recA, dnaK, and hsp60 (also known as groel and cpn60) has been more suitable to discriminate species for some groups [19, 43, 49, 70, 74, 95, 113]. An example is the use of the rpoB gene sequence for rapid identification of Bacillus and the use of hsp60 for phylogenetic and taxonomic studies of Enterobacter [49, 50, 56].
Multilocus sequence typing (MLST) is a new approach that has been used to obtain genetic information for characterization of distinct strains of bacteria within known species, analyzing sequences of internal fragments of a set of housekeeping loci (∼7). MLST has been considered a powerful tool for bacterial typing and has been developed for several pathogenic bacteria, such as Vibrio sp., Neisseria meningitides, Streptoccocus pyogenes, Staphylococus aureus and Campylobacter jejuni [10, 22, 30, 33, 37, 113]. The combination of allele numbers leads to the discovery of the sequence type. Feil et al. [33] concluded that variation at a single nucleotide site within a meningococcal housekeeping gene is more probable to occur as a result of recombination than due to point mutation in bound N. meningitidis clonal complexes. Thompson et al. [113] believes that MLST will improve the knowledge of taxonomy, and phylogeny of Vibrios indicated that MLST will improve knowledge on the taxonomy and phylogeny of Vibrios. MLST has been shown to be a very specific and unambiguous method for the characterization of bacteria species; recently, it was also used for non-pathogenic bacteria, including Lactobacillus plantarum [22].
Multilocus sequence analysis (MLSA) is a more general technique than MLST, suitable for genotypic characterization of a more diverse group, using sequences of genes that encode proteins, which are ubiquitous and are present in a single copy of the genome and present at least in the taxa under study. Bacterial identification is first made by sequencing the 16S rRNA gene of an unknown strain, identifying it at the genus or family level and then determining a set of genes and primers to be used to identify strains at the species level using MLSA [37]. The MLSA approach has been used to identify clinical and environmental enterococcal species, using rpoA and pheS genes; it is an efficient screening method for the detection of novel species [83].
Specific functional genes are other options for bacterial diversity studies, whenever metabolic function is known for cultured microorganisms. These genes are examined to determine the relation between structure and function, such as the genes pmoA and mxaF for methanotrophic bacteria [6, 53] and nodD for Rhizobium [120]. Cloning and sequencing of functional genes from environmental samples is extremely useful for investigation of ecologically distinct groups and classification at the species level [88, 89].
Importance of culturing techniques
Traditionally, culturing recovers only a minute fraction of bacterial diversity. This significantly reduces our understanding of the actual physiological and metabolic properties of the community of microorganisms found in natural environments [81, 92].
There is no doubt that the culture methodologies are still important tools for the study of prokaryote diversity. Continued research is required and new methods need to be developed for the isolation of newly identified organisms that are discovered through techniques that do not depend on bacterial culture. Culture is still needed to help understand the characteristics and properties of these groups and their contribution to the immense existing prokaryotic diversity [20, 54, 81, 97, 103].
Bacterial populations that are phylogenetically closely related can be distinct physiologically (definition of microdiversity), while phylogenetically distant species can have physiological similarities and can coexist in the same natural environment [60, 99].
Though we can obtain phylogenetic data about unculturable microbes isolated from natural environments, normally there is no means of obtaining information about their genetic and ecophysiological characteristics. However, the data obtained through molecular methodologies using 16S rRNA gene sequences are very important for our efforts to develop and improve culture methods for bacteria isolation that will allow studies of morphology, physiology, abundance, and distribution in natural habitats [60, 99]. Nowadays, some progress has been made in this area, in the development of new methods of culture that allow the isolation of bacteria previously not cultivable, in order to explore the potential of these microorganisms and to understand their ecological role. These studies have associated new culture techniques with analysis of 16S rRNA gene sequences, opening new perspectives and improving our knowledge about bacterial communities in many ecosystems [27, 29, 46, 59, 63, 76, 85, 92, 107]. When these methodologies are associated, most of the 16S rRNA gene sequences analyzed from clones of libraries constructed from total DNA extracted directly from environmental samples are found to be different from sequences obtained from cultivable bacteria isolated from the same sample, identical sequences rarely occurring [29, 92]. However, 16S rRNA gene sequence libraries have more objective data than those obtained with culture techniques. The combination of the two methodologies will likely provide additional information about the diversity and physiology of the natural bacterial community, giving relevance to the results obtained with the polyphasic approach [26, 27, 29, 76, 92]. Research with cultured bacteria that have been isolated is of enormous relevance for our knowledge of this practically unexplored universe, since bacteria perform essential roles in various ecosystems.
Metagenome: a function and sequence-driven approach
Nowadays, one of the most widely adopted strategies for studying microbial diversity is the metagenomic research. A metagenome is the entire genetic composition of microbial communities of an environment; this approach is based on direct isolation of total DNA in environmental samples, construction of libraries and the amplification of 16S rRNA genes and functional genes to study the total diversity, physiology, ecology and phylogeny of bacteria that cannot be cultivated in the laboratory (Fig. 2) [71, 100, 110, 111, 118]. Such investigations aim to reveal and understand the relationship between community composition and functional diversity in natural microbial ecosystems.

Scheme of construction and screening of environmental metagenomic libraries
Metagenomic research is useful to exploit the unknown bacterial diversity in different environments; it can be used to discover novel genes and to increase our knowledge on bacterial ecology and physiology [17, 110]. The 16S rRNA gene accounts for a minor fraction of the average prokaryotic genome and it does not give information about the physiology of the bacteria [97]. Metagenomic approaches using 16S rRNA gene sequences as a phylogenetic marker have been used to characterize uncultivated prokaryotes and can help to discover metabolic functions, enhancing our knowledge about bacterial ecology and phylogeny [86, 96, 110, 115].
We can use metagenomic sequences to help understand how complex microbial communities function and how bacteria interact within these niches. The diverse bacteria in a natural environment can be a complex chemical source of many undiscovered biodynamic compounds, with potential for bioprospecting. New antibiotics, enzymes and proteins have been identified by functional analysis of metagenomic libraries, including turbomycin A and B, lipases, amylases, nucleases and hemolytic activities (Table 3) [39, 96, 100]. Metagenomics thereby has two main goals: the identifying novel genes and increasing our understanding of microbial ecology [71]. This approach is very promising for novel biochemical and ecological discoveries, though development and improvement of new methodologies is essential for us to take advantage of this data.
Futures perspectives
All of the approaches that are available today have advantages and limitations, though none of them provide complete access to the extremely important and complex bacterial world. These new techniques, which are in constant development, have provided powerful and important confirmation of previous phenotypic and genotypic studies of bacteria. The combination of different methods is still the most suitable way of having a better understanding about diversity, phylogeny, ecology, evolution, and taxonomy of the largest group of living organisms on Earth—the Prokaryotes. Several questions remain to be resolved and the collaboration of taxonomists, microbiologists, and molecular biologists is essential and very important for the integration of the different research methods to allow for a proper assessment of microbial diversity and its real potential.
References
Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF (2004) Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol 186:2629–2635
Amann R, Fuchs BM, Behrens S (2001) The identification of microorganisms by fluorescence in situ hybridization. Curr Opin Biotechnol 12:231–236
Arber W (2000) Genetic variation: molecular mechanisms and impact on microbial evolution. FEMS Microbiol Rev 24:1–7
Baker GC, Smith JJ, Cowan DA (2003) Review and re-analysis of domain-specific 16S primers. J Microbiol Methods 55:541–555
Bernardet JF, Vancanneyt M, Matte-Taillieza O, Grisez L, Tailliez P, Bizet C, Nowakowski M, Kerouault B, Swings J (2005) Polyphasic study of Chryseobacterium strains isolated from diseased aquatic animals. Syst Appl Microbiol 28:640–660
Bodrossy L, Holmes EM, Holmes AJ, Kovács KL, Murrell JC (1997) Analysis of 16S rRNA and methane monooxygenase gene sequences reveals a novel group of thermotolerant and thermophilic methanotrophs, Methylocaldum gen. nov. Arch Microbiol 168:493–503
Brady SF, Clardy J (2004) Palmitoylputrescine, an antibiotic isolated from the heterologous expression. J Nat Prod 67:1283–1286
Brümmer IHM, Felske A, Wagner-Döbler I (2003) Diversity and seasonal variability of β-Proteobacteria in biofilms of polluted rivers: analysis by temperature gradient gel electrophoresis and cloning. Appl Environ Microbiol 69:4463–4473
Bull AT, Ward AC, Goodfellow M (2000) Search and discovery strategies for biotechnology: the paradigm shift. Microbiol Mol Biol Rev 64:573–606
Chan M, Maiden MCJ, Spratt BG (2001) Database-driven multilocus sequence typing (MLST) of bacterial pathogens. Bioinformatics 17:1077–1083
Cho J, Tiedje JM (2001) Bacterial species determination from DNA-DNA hybridization by using genome fragments and DNA microarrays. Appl Environ Microbiol 67:3677–3682
Christensen JJ, Kilian M, Fussing V, Andresen J, Blom J, Korner B (2005) Aerococcus urinae: polyphasic characterization of the species. APMIS 113:517–525
Chun J, Hug A, Colwell RR (1999) Analysis of 16S-23S rRNA intergenic spacer regions of Vibrio cholerae and Vibrio mimicus. Appl Environ Microbiol 65:2202–2208
Cohan FM (2001) Bacterial species and speciation. Syst Biol 50:513–524
Colwell RR (1970) Polyphasic taxonomy of the genus Vibrio: numerical taxonomy of Vibrio cholerae, Vibrio parahaemolyticus, and related Vibrio species. J Bacteriol 104:410–433
Cottrell MT, Moore JA, Kirchman DL (1999) Chitinases from uncultured marine microorganisms. Appl Environ Microbiol 65:2553–2557
Cowan D, Meyer Q, Stafford W, Muyanga S, Cameron R, Wittwer P (2005) Metagenomic gene discovery: past, present and future. Trends Biotechnol 23:321–329
Crosa JH, Brenner DJ, Falkow S (1973) Use of a single-strand specific nuclease for analysis of bacterial and plasmid deoxyribonucleic acid homo- and heteroduplexes. J Bacteriol 115:904–911
Dahllöf I, Baillie H, Kjelleberg S (2000) rpoB-based microbial community analysis avoids limitations inherent in 16S rRNA gene intraspecies heterogeneity. Appl Environ Microbiol 66:3376–3380
Dahllöf I (2002) Molecular community analysis of microbial diversity. Curr Opin Biotechnol 13:213–217
Dar SA, Kuenen JG, Muyzer G (2005) Nested PCR-denaturing gradient gel electrophoresis approach to determine the diversity of sulfate-reducing bacteria in complex microbial communities. Appl Environ Microbiol 71:2325–2330
de las Rivas B, Marcobal A, Muñoz R (2006) Development of a multilocus sequence typing method for analysis of Lactobacillus plantarum strains. Microbiol 152:85–93
Demain AL (2006) From natural products discovery to commercialization: a success story. J Ind Microbiol Biotechnol 33:486–495
Deutschbauer AM, Chivian D, Arkin AP (2006) Genomics for environmental microbiology. Curr Opin Biotechnol 17:1–7
Dolzani L, Tonin E, Lagatolla C, Monti-Bragadin C (1994) Typing of Staphylococcus aureus by amplification of the 16S-23S intergenic spacer sequences. FEMS Microbiol Lett 119:167–174
Donachie SP, Hou S, Lee KS, Riley CW, Pikina A, Belishe C, Kempe S, Gregory TS, Bossuyt A, Boerema J, Liu J, Freitas TA, Malahoff A, Alam M (2004) The Hawaiian archipelago: a microbial diversity hotspot. Microbiol Ecol 48:509–520
Dunbar J, Takala S, Barns SM, Davis JA, Kuske CR (1999) Levels of bacterial community diversity in four arid soils compared by cultivation and 16S rRNA gene cloning. Appl Environ Microbiol 65:1662–1669
Eardly BD, Nour SM, van Berkum P, Selander RK (2005) Rhizobial 16S rRNA and dnaK genes: mosaicism and the uncertain phylogenetic placement of Rhizobium galegae. Appl Environ Microbiol 71:1328–1335
Eilers H, Pernthaler J, Glöckner FO, Amann R (2000) Culturability and in situ abundance of pelagic bacteria from the North Sea. Appl Environ Microbiol 66:3044–3051
Enright MC, Spratt BG, Kalia A, Cross JH, Bessen D (2001) Multilocus sequence typing of Streptococcus pyogenes and the relationships between emm type and clone. Infect Immun 69:2416–2427
Entcheva P, Liebl W, Johann A, Hartsch T, Streit W (2001) Direct cloning from enrichment cultures, a reliable strategy for isolation of complete operons and genes from microbial consortia. Appl Environ Microbiol 67:89–99
Euzéby JP (1997) List of bacterial names with standing in nomenclature: a folder available on the internet. Int J Syst Bacteriol 47:590–592
Feil EJ, Maiden MCJ, Achtman M, Spratt BG (1999) The relative contributions of recombination and mutation to the divergence of clones of Neisseria meningitides. Mol Biol Evol 16:1496–1502
Fogel GB, Collins CR, Li J, Brunk CF (1999) Prokaryotic genome size and SSU rDNA copy number: estimation of microbial relative abundance from a mixed population. Microbiol Ecol 38:93–113
Forney LJ, Zhou X, Brown CJ (2004) Molecular microbial ecology: land of the one-eyed king. Curr Opin Microbiol 7:210–220
Fry J (2000) Bacterial diversity and ‘unculturables’. Microbiol Today 27:186–188
Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, Feil EJ, Stackebrandt E, van de Peer Y, Vandamme P, Thompson FL, Swings J (2005) Re-evaluating prokaryotic species. Nat Rev Microbiol 3:733–739
Gich FB, Amer E, Figueras JB, Abella CA, Balaguer MD, Poch M (2000) Assessment of microbial community structure changes by amplified ribosomal DNA restriction analysis (ARDRA). Int Microbiol 3:103–106
Gillespie DE, Brady SF, Bettermann AD, Cianciotto NP, Liles MR, Rondon MR, Clardy J, Goodman RM, Handelsman J (2002) Isolation of antibiotics turbomycin A and B from a metagenomic library of soil microbial DNA. Appl Environ Microbiol 68:4301–4306
Gillman LM, Gunton J, Turenne CY, Wolfe J, Kabani AM (2001) Identification of Mycobacterium species by multiple-fluorescence PCR–single-strand conformation polymorphism analysis of the 16S rRNA gene. J Clin Microbiol 39:3085–3091
Giraffa G, Paris A, Valcavi L, Gatti M, Neviani E (2001) Genotypic and phenotypic heterogeneity of Streptococcus thermophilus strains isolated from dairy products. J Appl Microbiol 91:937–943
Guerra B, Junker E, Schroeter A, Malorny B, Lehmann S, Helmuth R (2003) Phenotypic and genotypic characterization of antimicrobial resistance in German Escherichia coli isolates from cattle, swine and poultry. J Antimicrob Chemother 52:489–492
Gupta RS (2000) The phylogeny of proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol Rev 24:367–402
Hacene H, Rafa F, Chebhouni N, Boutaiba S, Bhatnagar T, Baratti JC, Ollivier B (2004) Biodiversity of prokaryoticmic microflora in El Golea Salt lake, Algerian Sahara. J Arid Environ 58:273–284
Hanage WP, Fraser F, Spratt BG (2005) Fuzzy species among recombinogenic bacteria. BMC Biol 3:1–7
Hanh MW (2004) Broad diversity of viable bacteria in “sterile” (0.2 mm) filtered water. Res Microbiol 155:688–691
Hassen A, Saidi N, Cherif M, Boudabous A (1998) Resistance of environmental bacteria to heavy metals. Bioresour Technol 64:7–15
Healy FG, Ray RM, Aldrich HC, Wilkie AC, Ingram LO, Shanmugam KT (1995) Direct isolation of functional genes encoding cellulases from the microbial consortia in a thermophilic, anaerobic digester maintained on lignocellulose. Appl Environ Microbiol 43:667–674
Hoffmann H, Roggenkamp A (2003) Population genetics of the nomenspecies enterobacter cloacae. Appl Environ Microbiol 69:5306–5318
Hoffmann H, Stindl S, Stumpf A, Mehlen A, Monget D, Heesemann J, Schleifer KH, Roggenkamp A (2005) Description of Enterobacter ludwigii sp. nov., a novel Enterobacter species of clinical relevance. Syst Appl Microbiol 28:206–212
Hongoh Y, Yuzawa H, Ohkuma M, Kudo T (2003) Evaluation of primers and PCR conditions for the analysis of 16S rRNA genes from a natural environment. FEMS Microbiol Lett 221:299–304
Horner-Devine MC, Carney KM, Bohannan BJM (2004) An ecological perspective on bacterial biodiversity. Proc R Soc Lond 271:113–122
Horz H, Yimga MT, Liesack W (2001) Detection of methanotroph diversity on roots of submerged rice plants by molecular retrieval of pmoA, mmoX, mxaF, and 16S rRNA and ribosomal DNA, including pmoA-based terminal restriction fragment length polymorphism profiling. Appl Environ Microbiol 67:4177–4185
Hugenholtz P, Goebel B, Pace NR (1998a) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Hugenholtz P, Pitulle C, Hershberger KL, Pace NR (1998b) Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol 180:366–376
Iversen C, Waddington M, On SLW, Forsythe S (2004) Identification and phylogeny of Enterobacter sakazakii relative to Enterobacter and Citrobacter species. J Clin Microbiol 42:5368–5370
Iwamoto T, Nasu M (2001) Current bioremediation practice and perspective. J Biosci Bioeng 92:1–8
Janda JM, Abbott SL (2002) Bacterial identification for publication: when is enough enough? J Clin Microbiol 40:1887–1891
Jaspers E, Nauhaus, Cypionka H, Overmann J (2001) Multitude and temporal variability of ecological niches as indicated by the diversity of cultivated bacterioplankton. FEMS Microbiol Ecol 36:153–164
Jaspers E, Overmann J (2004) Ecological significance of microdiversity: identical 16S rRNA gene sequences can be found in bacteria with highly divergent genomes and ecophysiologies. Appl Environ Microbiol 70:4831–4839
Jensen MA, Webster JA, Straus N (1993) Rapid identification of bacteria on the basis of polymerase chain reaction-amplified ribosomal DNA spacer polymorphisms. Appl Environ Microbiol 59:945–952
Kaeberlein T, Lewis K, Epstein SS (2002) Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296:1127–1129
Keller M, Zengler K (2004) Tapping into microbial diversity. Nat Rev Microbiol 2:141–150
Kemp PF, Aller JY (2004) Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiol Ecol 47:161–177
Klappenbach JA, Saxman PR, Cole JR, Schmidt TM (2001) rrndb: the ribosomal RNA operon copy number database. Nucleic Acids Res 29:181–184
Knietsch A, Waschkowitz T, Bowien S, Henne A, Daniel R (2003) Construction and screening of metagenomic libraries derived from enrichment cultures: generation of a gene bank for genes conferring alcohol oxidoreductase activity on Escherichia coli. Appl Environ Microbiol 69:1408–1416
Lebuhn M, Bathe S, Achouak W, Hartmann A, Heulin T, Schloter M (2006) Comparative sequence analysis of the internal transcribed spacer 1 of Ochrobactrum species. Syst Appl Microbiol 29:265–275
Lagacé L, Pitre M, Jacques M, Roy D (2004) Identification of the bacterial community of maple sap by using amplified ribosomal DNA (rDNA) restriction analysis and rDNA sequencing. Appl Environ Microbiol 70:2052–2060
Levin BR, Bergstrom CT (2000) Bacteria are different: observations, interpretations, speculations, and opinions about the mechanisms of adaptive evolution in prokaryotes. PNAS 97:6981–6985
López-López A, Bartual SG, Stal L, Onyshchenko O, Rodríguez-Valera F (2005) Genetic analysis of housekeeping genes reveals a deep-sea ecotype of Alteromonas macleodiiin the Mediterranean Sea. Environ Microbiol 7:649–659
Lorenz P, Schleper C (2002) Metagenome—a challenging source of enzyme discovery. Mol Catal B Enzym 19–20:13–19
Lyautey E, Lacoste B, Ten-Hage L, Rols J, Garabetian F (2005) Analysis of bacterial diversity in river biofilms using 16S rDNA PCR-DGGE: methodological settings and fingerprints interpretation. Water Res 39:380–388
Marshall S, Melito PL, Woodward DL, Johnson WM, Rodgers FG, Mulvey M (1999) Rapid identification of Campylobacter, Arcobacter, and Helicobacter isolates by PCR-Restriction Fragment Length Polymorphism analysis of the 16S rRNA gene. J Clin Microbiol 37:4158–4160
Masson L, Maynard C, Brousseau R, Goh S, Hemmingsen SM, Hill JE, Paccagnella A, Oda R, Kimura N (2006) Identification of pathogenic Helicobacter species by chaperonin-60 differentiation on plastic DNA arrays. Genomics 87:104–112
Matsuda M, Tazumi A, Kagawa S, Sekizuka T, Murayama O, Moore JE, Millar BC (2006) Homogeneity of the 16S rDNA sequence among geographically disparate isolates of Taylorella equigenitalis. BMC Vet Res 2:1–4
McCaig AE, Grayston SJ, Prosser JI, Glover A (2001) Impact of cultivation on characterisation of species composition of soil bacterial communities. FEMS Microbiol Ecol 35:37–48
McInerney JO, Wernecke M, Mullarkey M, Powell R (2002) Bacteria and Archaea: molecular techniques reveal astonishing novel diversity. Biodiversity 3:3–10
McMullan G, Christie JM, Rahman TJ, Banat IM, Ternan NG, Marchant R (2004) Habitat, applications and genomics of the aerobic, thermophilic genus Geobacillus. Biochem Soc Trans 32:214–217
Mechichi T, Stackebrandt E, Gadon N, Fuchs G (2002) Phylogenetic and metabolic diversity of bacteria degrading aromatic compounds under denitrifying conditions, and description of Thauera phenylaceticasp. nov., Thauera aminoaromatica sp. nov., and Azoarcus buckeliisp. nov. Arch Microbiol 178:26–35
Mehlen A, Goeldner M, Ried S, Stindl S, Ludwig W, Schleifer K (2004) Development of a fast DNA-DNA hybridization method based on melting profiles in microplates. Syst Appl Microbiol 27:689–695
Miteva VI, Scheridant PP, Brenchley JE (2004) Phylogenetic and physiological diversity of microorganisms isolated from a deep Greenland glacier ice core. Appl Environ Microbiol 70:202–213
Morris CE, Bardin M, Berge O, Frey-Klett P, Fromin N, Girardin H, Guinebretière MH, Lebaron P, Thiéry JM, Troussellier M (2002) Microbial biodiversity: approaches to experimental design and hypothesis testing in primary scientific literature from 1975 to 1999. Microbiol Mol Biol Rev 66:592–616
Naser SM, Thompson FL, Hoste B, Gevers D, Dawyndt D, Vancanneyt M, Swingers J (2005) Application of multilocus sequence analysis (MLSA) for rapid identification of Enterococcus species based on rpoA and pheS genes. Microbiol 151:2141–2150
Nee S (2003) Unveiling prokaryotic diversity. Trends Ecol Evol 18:62–63
O’Sullivan LA, Fuller KE, Thomas EM, Turley CM, Fry JC, Weightman AJ (2004) Distribution and culturability of the uncultivated ‘AGG58 cluster’ of the Bacteroidetes phylum in aquatic environments. FEMS Microbiol Ecol 47:359–370
Oremland RS, Capone DG, Stolz JF, Fuhrman J (2005) Whither or wither geomicrobiology in the era of ‘community metagenomics’. Nat Rev Microbiol 3:572–578
Oren A (2004) Prokaryote diversity and taxonomy: current status and future challenges. Philol Trans R Soc Lond 359:623–638
Palumbo AV, Schryver JC, Fields MW, Bagwell CE, Zhou JZ, Yan T, Liu X, Brandt CC (2004) Coupling of functional gene diversity and geochemical data from environmental samples. Appl Environ Microbiol 70:6525–6534
Palys T, Berger E, Mitrica I, Nakamura LK, Cohan FM (2000) Protein-coding genes as molecular markers for ecologically distinct populations: the case of two Bacillus species. Int J Syst Evol Microbiol 50:1021–1028
Palys T, Nakamura LK, Cohan FM (1997) Discovery and classification of ecological diversity in the bacterial world: the role of DNA sequence data. Int J Syst Bacteriol 47:1145–1156
Papamanoli E, Kotzekidou P, Tzanetakis N, Litopoulou-Tzanetaki E (2002) Characterization of Micrococcaceae isolated from dry fermented sausage. Food Microbiol 19:441–449
Pearce DA, van der Gast CJ, Lawley B, Ellis-Evans JC (2003) Bacterioplankton community diversity in a maritime Antarctic lake, determined by culture-dependent and culture-independent techniques. FEMS Microbiol Ecol 45:59–70
Perreten V, Vorlet-Fawer L, Slickers P, Ehricht R, Kuhnert P, Frey J (2005) Microarray-based detection of 90 antibiotic resistance genes of gram-positive bacteria. J Clin Microbiol 43:2291–2302
Pretzer G, Snel J, Molenaar D, Wiersma A, Bron PA, Lambert J, de Vos VM, van der Meer R, Smits MA, Kleerebezem M (2005) Biodiversity-based identification and functional characterization of the mannose-specific adhesin of Lactobacillus plantarum. J Bacteriol 187:6128–6136
QI Y, Patra G, Liang X, Williams LE, Rose S, Redkar RJ, Delvecchio VG (2001) Utilization of the rpoB gene as a specific chromosomal marker for real-time PCR detection of Bacillus anthracis. Appl Environ Microbiol 67:3720–3727
Riesenfeld CS, Schloss PD, Handelsman J (2004) Metagenomics: genomic analysis of microbial communities. Annu Rev Genet 38:525–552
Rodríguez-Valera F (2002) Approaches to prokaryotic biodiversity: a population genetics perspective. Environ Microbiol 4:628–633
Rodríguez-Valera F (2004) Environmental genomics, the big picture? FEMS Microbiol Lett 231:153–158
Rosselló-Mora R, Amann R (2001) The species concept for prokaryotes. FEMS Microbiol Rev 25:39–67
Rondon MR, August PR, Bettermann AD, Brady SF, Grossman TH, Liles MR, Loiacono KA, Lynch BA, MacNeil IA, Minor C, Tiong CL, Gilman M, Osburne MS, Clardy J, Handelsman J, Goodman RM (2000) Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol 66:2541–2547
Santosa DA (2001) Rapid extraction and purification of environmental DNA for molecular cloning applications and molecular diversitym studies. Mol Biotechnol 17:59–64
Schirmer A, Gadkari R, Reeves CD, Ibrahim F, DeLong EF, Hutchinson CR (2005) Metagenomic analysis reveals diverse polyketide synthase gene clusters in microorganisms associated with the marine sponge Discodermia dissolute. Appl Environ Microbiol 71:4840–4849
Schleifer K (2004) Microbial diversity: facts, problems and prospects. System Appl Microbiol 27:3–9
Schloss PD, Handelsman J (2004) Status of the microbial census. Microbiol Mol Biol Rev 68:686–691
Schmalenberger A, Schwieger F, Tebbe CC (2001) Effect of primers hybridizing to different evolutionarily conserved regions of the small-subunit rRNA gene in PCR-based microbial community analyses and genetic profiling. Appl Environ Microbiol 67:3557–3563
Sievert SM, Kuever J, Muyzer G (2000) Identification of 16S ribosomal DNA-defined bacterial populations at a shallow submarine hydrothermal vent near Milos Island (Greece). Appl Environ Microbiol 66:3102–3109
Stabili L, Cavallo RA (2004) Biodiversity of culturable heterotrophic bacteria in the southern adriatic sea Italian coastal waters. Sci Mar 68:31–41
Stackebrandt E, Frederiksen W, Garrity GM, Grimont PAD, Kämpfer P, Maiden MCJ, Nesme X, Rosselló-Mora R, Swings J, Trüpper HG, Vauterin L, Ward AC, Whitman WB (2002) Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol 52:1043–1047
Staley JT, Krieg NR (1989) Classification of prokaryotic organisms: an overwiew. In: Williams ST, Sharpe ME, Holtz JG (eds) Bergey’s manual of systematic bacteriology, vol4.. Williams and Wilkins, Baltimore, MD, pp 2299–2306
Steele HL, Streit WR (2005) Metagenomics: advances in ecology and biotechnology. FEMS Microbiol Lett 247:105–111
Streit WR, Schmitz RA (2004) Metagenomics—the key to the uncultured microbes. Curr Opin Microbiol 7:492–498
Teusink B, Smid EJ (2006) Modelling strategies for the industrial exploitation of lactic acid bacteria. Nature 4:46–56
Thompson FL, Iida T, Swings J (2004) Biodiversity of Vibrios. Microbiol Mol Biol Res 68:403–431
Torsvik V, Ovreas L, Thingstad TF (2002) Prokaryotic diversity—magnitude, dynamics, and controlling factors. Science 296:1064–1066
Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, Bork P, Hugenholtz P, Rubin EM (2005) Comparative metagenomics of microbial communities. Science 308:554–557
van Berkum P, Terefework Z, Paulin L, Suomalainen S, Lindström K, Eardly BD (2003) Discordant phylogenies within the rrn loci of Rhizobia. J Bacteriol 185:2988–2998
Vandamme P, Pot B, Gillis M, De vos P, Kersters K, Swings J (1996) Polyphasic taxonomy, a consensus approach to bacterial systematics. Microbiol Rev 60:407–438
Voget S, Leggewie C, Uesbeck A, Raasch C, Jaeger KE, Streit WR (2003) Prospecting for novel biocatalysts in a soil metagenome. Appl Environ Microbiol 69:6235–6242
Zeigler DR (2003) Gene sequences useful for predicting relatedness of whole genomes in bacteria. Int J Syst Evol Microbiol 53:1893–1900
Zézé A, Mutch LA, Young JPW (2001) Direct amplification of nodD from community DNA reveals the genetic diversity of Rhizobium leguminosarum in soil. Environ Microbiol 3:363–70
Ward DM (1998) A natural species concept for prokaryotes. Curr Opin Microbiol 1:271–277
Wayne LG, Brenner DJ, Colwell RR, Grimont PAD, Kandler O, Krichevsky MI, Moore LH, Morre WEC, Murray RGE, Stackebrandt E, Starr MP, Truper HG (1987) Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol 37:463–464
Wertz JE, Goldstone C, Gordon DM, Riley MA (2003) A molecular phylogeny of enteric bacteria and implications for a bacterial species concept. J Evol Biol 16:1236–1248
Whitman WB, Coleman DC, Wiebe WJ (1998) Prokaryotes: the unseen majority. Proc Natl Acad Sci 95:6578–6583
Woese CR (1987) Bacterial evolution. Microbiol Rev 51:221–271
Wu X, Walker MJ, Hornitzky M, Chin J (2005) Development of a group-specific PCR combined with ARDRA for the identification of Bacillus species of environmental significance. J Microbiol Methods 64:107–119
Acknowledgments
We appreciate the financial support given by PELD and CNPq in the form of a scholarship to Daniela Santos Pontes.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pontes, D.S., Lima-Bittencourt, C.I., Chartone-Souza, E. et al. Molecular approaches: advantages and artifacts in assessing bacterial diversity. J Ind Microbiol Biotechnol 34, 463–473 (2007). https://doi.org/10.1007/s10295-007-0219-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-007-0219-3