
Abstract
In most bacteria acetate assimilation is accomplished via the glyoxylate pathway. Isocitrate lyase (ICL) and malate synthase (MS) are two key enzymes of this pathway, which results in the net generation of one molecule of succinyl-CoA from two acetyl-CoA molecules. Genetic and biochemical data have shown that genes encoding these key enzymes are present in streptomycetes, yet there has been no clear demonstration of the importance of these genes to acetate assimilation. In fact, for Streptomyces collinus an alternative butyryl-CoA pathway has been shown to be critical for growth on acetate as a sole carbon source. Crotonyl-CoA reductase (CCR) is a key enzyme in this pathway and catalyzes the last step of the conversion of 2-acetyl-CoA molecules to butyryl-CoA. In Streptomyces cinnamonensis C730.1, it has been shown that CCR and this butyryl-CoA pathway provide the majority of methylmalonyl-CoA and ethylmalonyl-CoA for monensin A biosynthesis in an oil-based fermentation medium. We have cloned a MS homologue gene from this strain. Reverse transcription and direct enzyme assays demonstrated that neither this nor other MS genes were expressed during fermentation in an oil-based fermentation of either the C730.1 or L1 strain (a ccr mutant). Similarly, no ICL activity could be detected. The C730.1 but not the L1 strain was able to grow on acetate as a sole carbon source. The Streptomyces coelicolor aceA and aceB2 genes encoding ICL and MS were cloned into a Streptomyces expression plasmid (a derivative of pSET152) to create pExIM1. Enzyme assays and transcript analyses demonstrated expression of both of these proteins in C730.1/pExIM1 and L1/pExIM1 grown in an oil-based fermentation and tryptic soy broth media. Nonetheless, L1/pExIM1, like L1, was unable to grow on acetate as a sole carbon source, and was unable to efficiently generate precursors for monensin A biosynthesis in an oil-based fermentation, indicating that the additional presence of these two enzyme activities does not permit a functional glyoxylate cycle to occur. UV mutagenesis of S. cinnamonensis L1 and L1/pExIM1 led to mutants which were able to grow efficiently on acetate despite a block in the butyryl-CoA pathway. Analysis of enzyme activity and monensin production from these mutants in an oil-based fermentation demonstrated that neither the glyoxylate cycle nor the butyryl-CoA pathway function, suggesting the possibility of alternative pathways of acetate assimilation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
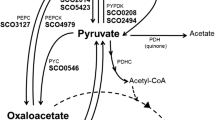
Aerobic microorganisms, which can grow on acetate, or other compounds which enter central metabolism at the level of acetyl-CoA, generally use the glyoxylate pathway. The key enzymes in this pathway are isocitrate lyase (ICL), cleaving isocitrate to succinate and glyoxylate, and malate synthase (MS), condensing glyoxylate and acetyl-CoA to form malate and CoA (Fig. 1a) [4]. These two enzymes in combination with enzymes from the Krebs cycle catalyze the net formation of succinyl-CoA from two acetyl-CoA molecules. Putative MS genes (aceB1 and aceB2) have been identified in Streptomyces coelicolor [1, 13] and the aceB2 gene product has been expressed in E. coli and shown to catalyze the predicted formation of malate from glyoxylate and acetyl-CoA [1, 13, 15]. This activity has also been observed for the protein product of an aceB gene, cloned from Streptomyces clavuligerus, S. griseus, and S. arenae, and expressed in Escherichia coli [6, 13, 15]. The ICL gene (aceA) has also been identified in both S. clavuligerus and S. coelicolor [20]. For S. coelicolor aceA is clustered with aceB2, separated by only 100 nt [13] (Fig. 2). A second S. coelicolor MS gene, aceB1, is clustered with an iclR homologue, an important transcriptional regulator for the glyoxylate pathway (Fig. 2) (a similar organization of iclR and aceB1 is found in Streptomyces avermitilis) [7]. The aceA from S. clavuligerus has been expressed and shown to have the predicted catalytic activity. Despite these observations there is no evidence that a functional glyoxylate pathway permits growth of either of these streptomycetes on acetate as a sole carbon source. In the case of S. clavuligerus growth on acetate (0.5% w/v) is delayed and high levels of ICL and MS coincide with cessation of growth, suggesting a late and as yet undetermined physiological role [2, 20].

Pathways for acetate assimilation. a The glyoxylate cycle. b The butyryl-CoA pathway. c Citramalate cycle for acetate assimilation as proposed in reference 8. ICL Isocitrate lyase, CMS citramalate synthase), MMC β-methylmalyl-CoA lyase, MS malate synthase, ML malyl-CoA lyase, MCL malyl-CoA lyase. Source: Akopiants et al.

Genetic architecture of aceB loci of Streptomyces and construction of the pExIM1 expression plasmid. Source: Akopiants et al.
An alternative butyryl-CoA pathway (Fig. 1b) was proposed as important for growth of S. collinus on acetate. In this process, the enzyme crotonyl-CoA reductase (CCR) plays a key role in catalyzing the last step in the formation of butyryl-CoA from two molecules of acetyl-CoA. The most direct route to succinyl-CoA then involves isomerization to isobutyryl-CoA, oxidation to methylmalonyl-CoA and a second isomerization step catalyzed by methylmalonyl-CoA mutase [4, 21]. A ccr mutant of S. collinus has been reported to be blocked in growth on acetate [4]. The ccr gene lies in a cluster of other genes including meaA (encoding a putative coenzyme B12-dependent mutase) and mcl1. There is evidence that it is this set of genes which is important for acetate metabolism in both S. collinus and Methylobacterium extorquens. In the ICL-negative M. extorquens a series of genetic experiments have led to the suggestion that CCR also catalyzes the final step in formation of butyryl-CoA from acetyl-CoA, while MeaA is proposed to play a role in a more circuitous route leading to methylmalonyl-CoA and succinyl-CoA [10]. In S. collinus MeaA also appears to be important for growth on acetate and has been implicated in a pathway that provides methylmalonyl-CoA. Nonetheless, the catalytic role of MeaA in both of these organisms remains undetermined. The mcl1 encodes a homologue of malyl-CoA lyase (MCL) in the Rhodobacter capsulatus, an ICL-negative bacterium. In this case, the mcl1 gene product has both l-malyl-CoA and β-methylmalyl-CoA lyase activity and is proposed to catalyze two steps in the citramalate cycle (Fig. 1c), an alternative pathway for acetate assimilation. MCL catalyzes the reversible condensation of glyoxylate and acetyl-CoA, to form malyl-CoA. In combination with a suitable MCL activity it can perform the same catalytic role as a MS. The mcl1 homologue is not only present in S. collinus but also in S. coelicolor, and clustered with ccr and meaA.
The butyryl-CoA pathway (Fig. 1b) also appears to play an important role in providing ethylmalonyl-CoA and methylmalonyl-CoA precursors for polyketide biosynthesis. Streptomyces cinnamonensis C730.1 produces the polyether antibiotic monensin A from seven methylmalonyl-CoA, one ethylmalonyl, and five malonyl-CoA units. The monensin B analog does not use ethylmalonyl-CoA but rather eight methylmalonyl-CoA precursors. The S. cinnamonensis ccr gene has been cloned and sequenced, and a ccr mutant strain (L1) constructed [12]. In glucose-soybean fermentation the L1 strain generates the same monensin titers as the wild-type strain but significantly more monensin B [12]. This observation suggested that under these growth conditions the butyryl-CoA pathway was important for providing ethylmalonyl-CoA (formed by carboxylation of butyryl-CoA) for monensin A biosynthesis, but not in methylmalonyl-CoA. More recently it has been shown that in an oil-based extended fermentation medium, there is a dramatic decrease in the methylmalonyl-CoA pool and overall titers for the L1 mutant as compared to the parent strain [11]. This observation indicates that under these conditions where the major carbon source comes from lipid degradation and enters central metabolism at the level of acetyl-CoA, the butyryl-CoA pathway provides the major route to methylmalonyl-CoA.
In this work, we cloned an aceB homologue from S. cinnamonensis. We showed through transcript analysis that this gene was not expressed in S. cinnamonensis grown in an oil-based extended fermentation medium. Similarly, no aceA transcripts could be observed under these conditions and ICL activity was not detected in cell extracts. The L1 strain was unable to grow on acetate, whereas the parent S. cinnamonensis C730.1 strain could. These data support the role of a butyryl-CoA pathway and not the glyoxylate pathway for growth of this strain on acetate. The plasmid pExIM1 was constructed to give constitutive expression of the S. coelicolor aceA and aceB2 genes in the L1 strain, and was confirmed by both transcript analyses and ICL assays of L1/pExIM1 grown in an oil-based extended fermentation. Nonetheless, L1/pExIM1 was unable to grow on acetate, indicating that the presence of ICL and MS activities alone is not sufficient for a functional glyoxylate pathway. UV mutagenesis of the L1 strain provided mutants able to grow on acetate as a sole carbon source. Analyses of these mutants demonstrated that the CCR activity and ICL activity were still absent, indicating that growth on acetate is accomplished by some pathway other than the glyoxylate cycle or the butyryl-CoA pathway.
Materials and methods
Strains and plasmids
Streptomyces cinnamonensis C730.1 and S. cinnamonensis L1 have been described previously [12]. Escherichia coli DH5α strain was used as the standard host for cloning procedures. Plasmids pBAD TOPO TA and pCR 2.1 TOPO TA were purchased from Invitrogen. Streptomyces coelicolor BAC 19F3 containing ICL and MS genes was provided by Dr. Helen Kieser at the John Innes Centre, Norwich, UK.
Fermentation of S. cinnamonensis
Fermentation of S. cinnamonensis in an oil-based medium was as described previously [11]. Fermentation on glucose-based medium was performed as described previously [11]. Medium components were supplied by Difco and Fisher. All other chemicals were purchased from Sigma. Purification of monensin A and quantitation of monensins from the fermentation broth were performed as described previously [11, 18].
DNA isolation, amplification, manipulation
Standard molecular cloning techniques were employed [5, 19]. An S. cinnamonensis C730.1 genomic library was constructed using the Supercos 1 vector kit and Gigapack III Gold cloning kit (Stratagene, La Jolla, Calif.) according to the manufacturer’s recommendations. PCR reactions were performed using the GC-Rich PCR system (Roche, Penzberg, Germany) as suggested by the manufacturer. A list of primers used in this work is presented in Table 1.
Transcript analysis
In order to prevent mRNA degradation during isolation, two volumes of RNAprotect reagent (Qiagen, Hilden, Germany) were added to one volume of the broth. Preparation of total RNA was then accomplished using an RNAeasy Midi kit (Qiagen) following the manufacturer's protocol, with an additional step of treatment of the lysate with a saturated phenol chloroform solution. Nucleic acid preparations were treated with DNAse I (DNA-free kit, Ambion) as recommended by the manufacturer. Nucleic dotMETRIC kits from Genotechnology were used for RNA quantification. A one-step RT-PCR kit from Qiagen was used for RT-PCR with addition of dimethyl sulfoxide (5% v/v final) to the RT-PCR reaction mix. The RT-PCR reaction conditions were as follows: an initial DNA strand synthesis with reverse transcriptase, 52°C for 30 min, followed by heating at 95°C for 15 min to activate the DNA polymerase, and then 25 cycles of 94°C for 10 s, 55°C for 20 s and 72°C for 45 s. Negative controls were carried out with each experimental reaction, using the same enzyme mix but without the initial reverse transcription step. All RT-PCR products were cloned into the pCR 2.1 TOPO cloning vector and sequenced.
Construction of pExIM1 and pICL2
The plasmid pExIM1 (Fig. 2) was constructed to obtain constitutive expression of the S. coelicolor aceA and aceB2 genes in the S. cinnamonensis L1 strain. The aceA and aceB2 were amplified using DNA BAC 19 F3 from S. coelicolor as a template and sets of primers designed based on the sequence of the S. coelicolor genome (Table 1). The aceA was amplified using icl3 primers and the resulting 1280 nt fragment flanked by NdeI-XmaI sites was cloned into pCR2.1 TOPO TA cloning vector providing pICL1. The aceB2 was amplified using ms4 primers and the 1592 nt DNA fragment flanked by XmaI-EcoRV sites was cloned into the pCR2.1 TOPO TA cloning vector to provide plasmid pMS1. The XmaI-EcoRV fragment from pMS1 was then ligated with XmaI-EcoRV digested pICL1, giving pEx1. The 2890 nt DNA fragment from NdeI-BglII digested pEx1 was then ligated with the 7.0 kb NdeI-BglII digested fragment of pGF200 [11] giving pExIM1. The pGF200 is a derivative of a conjugative vector pSET152 with ermE promoter, carrying the ribosome-binding site from pET15b (the second RBS site 5′ of aceB2 in pExIM1was introduced by primer ms4). The pExIM1 plasmid was transferred to S. cinnamonensis C730 L1 by conjugation and ApR clones were selected.
The aceA gene in pExIM1 was amplified using the icl6 (see Table 1) primer pair and cloned into pBAD TOPO TA to provide pICL2 (for expression in E. coli).
Generation of UV mutants of S. cinnamonensis L1 able to grow on acetate
Mutants of S. cinnamonensis L1 were obtained and tested by standard procedure with only minor modifications. Irradiated spores (killing rate 99.99%) of both S. cinnamonensis L1, and L1/pExIM1 were plated directly onto minimal medium [5] with sodium acetate (5 g/l) as a carbon source. Colonies (M1-3 and MM1-3, respectively) were transferred to fresh plates with sodium acetate after 4 days of growth (conducted in the dark to prevent photoreactivation).
Ethyl [3,4-13C2]acetoacetate incorporation experiments with S. cinnamonensis L1, M1, and MM1
These experiments were carried out following methods described previously for the S. cinnamonensis L1 mutant [11].
Enzyme activity in crude extracts
Protein concentrations were determined using a Micro BCA protein assay kit (Pierce, Rockford, Ill.). ICL, and CCR enzyme assays in crude extracts of S. cinnamonensis C 730.1 and E. coli were carried out as described previously [4]. Malate dehydrogenase activity was determined using a spectrophotometric assay [17].
Results
Cloning and sequencing a MS gene (aceB) from S. cinnamonensis C730.1
MSs cloned from Streptomyces thus far share significant amino acid homology. For example, there is 83% amino acid identity between AceB2 S. coelicolor NP_62579.1 [1] and putative AceB2 MS S. avermitilis MA-4680 NP_823220.1 [16]. The AceB1 from these two organisms exhibit even higher amino acid identity (90%) and there is also significant amino acid identity between the AceB1 and AceB2 within and between the organisms (56–56%). We used this sequence identity to construct PCR primers (ms in Table 1) to test for the presence of MS genes in S. cinnamonensis C730.1. Using this primer set and total DNA of S. cinnamonensis C730.1 strain as a template, a 403 nt DNA fragment was amplified, cloned and sequenced. Sequence analysis of this fragment revealed >90% amino acid identity to known and putative MS genes aceB1 from S. avermitilis MA-4680 (NP_823176) and S. coelicolor. This DNA fragment was used to probe a library of S. cinnamonensis 730.1 genomic DNA and led to the identification of four clones with similar restriction profiles. A cosmid clone designated 3C1 was partially sequenced by primer walking, initially using primers complementary to ms. This complete aceB ORF, encoding a probable MS AceB in S. cinnamonensis C730.1, is 1619 nt long and has high nucleotide identity with MS aceB of S. griseus AAK14382.1 (91% identity) S. coelicolor aceB1 (90%), and S. avermitilis (89%).
The genetic background of the sequenced locus is similar to that of aceB1 of S. coelicolor but not of aceB2 (Fig. 2). The 3′ and 5′ flanking regions in S. coelicolor contain SCO6242 and SCO6244, respectively. A SCO6244 homologue is also located 5′ of the S. avermitilis aceB1. SCO6242 and SCO6244 homologues also flank the S. cinnamonensis aceB1. The iclR gene, encoding a regulator of the glyoxylate pathway is also located as the second ORF 5′ to aceB1 in S. coelicolor (SCO6246) and S. avermitilis MA-4680. Additional sequencing of the 3C1 cosmid clone would permit a determination of whether there is an iclR homologue 5′ of the S. cinnamonensis aceB. The S. cinnamonensis aceB gene was deposited in GenBank (accession number DQ003081).
Detection of ICL and MS in S. cinnamonensis strains
RT-PCR was used to detect the presence of aceA and aceB2 transcripts during fermentation of S. cinnamonensis in an oil-based fermentation medium. Using primers sets icl1 and ms2 and ms5 (Table 1) we could not detect either aceA or aceB transcripts in S. cinnamonensis 730.1 in an oil-based fermentation. The icl1 primer set was designed based on highly conserved regions of Streptomycete ICLs. The ms2 primer pair was designed based on highly conserved aceB regions, while the ms5 pair was designed based on the S. cinnamonensis aceB. The CCR (ccr) gene was used in RT-PCR experiments as a positive control (primers ccrc in Table 1) as we have previously shown that transcripts are present at all stages in this oil-based S. cinnamonensis fermentation. In the case of aceB, no RT-PCR products were obtained after 25 cycles, and the PCR products obtained with more than 30 cycles were shown to be a result of DNA contamination (lanes 1–6, Fig. 3). In contrast a clear RT-PCR product was obtained for ccr after 25 cycles (lane 8, Fig. 3). These data suggest that the S. cinnamonensis aceB gene was not expressed.

RT-PCR analysis of aceB expression in S. cinnamonensis C 730.1 during an oil-based extended fermentation. Templates are total RNA from strain S. cinnamonensis C730.1 after 48 h of fermentation in MOF medium. Reverse transcription with primers ms5 (see Table 1): lane 1 40 cycles, lane 3 45 cycles, lane 5 50 cycles. PCR control for DNA contamination with primers ms5 with same templates as for reverse transcription: lane 2 40 cycles, lane 4 45 cycles, lane 6 50 cycles. Lane 8 Reverse transcription for ccr gene primers ccrc with the same RNA template, 25 cycles; lane 9 PCR control for DNA contamination for ccr gene primers ccrc, 25 cycles; lanes 7 and 10 DNA ladders. Source: Akopiants et al.
In the case of aceA, no PCR products were obtained using either RT-PCR from RNA samples, or PCR from S. cinnamonensis C730.1 DNA (data not shown). Thus, it is not yet possible to determine if S. cinnamonensis contains an aceA gene. However, no ICL activity could be observed from cell extracts generated from S. cinnamonensis C730.1 growing in an oil-based fermentation medium (bar 2 in Fig. 4), suggesting that even if there is an aceA, it was not expressed to any significant level under these conditions.

Isocitrate lyase activity in cell extracts generated from S. cinnamonensis strains grown in oil-based extended fermentation. 1 L1 pExIM1, 2 C730.1, 3 L1, 4 MM1, 5 MM2, 6 MM3. Samples were taken after 24 h in MOF medium. Similar observations were made for samples taken at 48 and 90 h. Source: Akopiants et al.
It has been shown previously that when S. collinus is grown in minimal medium containing Tween as carbon source, ICL activity can be observed in cell extracts. Similar observations have been made for S. coelicolor [4]. We could not test if similar observations could be made for S. cinnamonensis C730.1 as we were unable to obtain conditions in which it would grow on Tween as a sole carbon source.
Expression of the S. coelicolor aceA and aceB2 genes in S. cinnamonensis
The plasmid pExIM1 (Fig. 2) was constructed to provide constitutive expression of the S. coelicolor aceA and aceB2 genes in the S. cinnamonensis L1 strain. The expression of these genes during fermentation in an oil-based medium was confirmed using RT-PCR (Fig. 5). Both aceA and aceB2 transcripts were detected using 15 cycles of RT-PCR (Fig. 5). Similarly, ICL activity was readily detected for S. cinnamonensis C730.1/pExIM1 grown in either oil-based (Fig. 4, bar 1) or tryptic soy broth (TSB) medium (data not shown). The S. coelicolor aceA gene was PCR-amplified from pExIM1 and cloned into the pBAD TOPO TA expression system (Invitrogen) to provide pICL2. Cell extracts of E. coli/pICL2 before arabinose induction demonstrated no detectable ICL activity whereas those generated 6 h after induction had very clear activity (Fig. 6). Taken together these assays of cell extracts of E. coli/pICl2 and S. cinnamonensis L1/pExIM1, provided the first clear biochemical evidence that the aceA identified by sequencing the S. coelicolor genome, encodes an AceA with ICL activity.

RT-PCR analysis of heterologous expression of the S. coelicolor aceA and aceB2 in S. cinnamonensis L1/pExIM1 in an oil-based fermentation. RT-PCR was carried out using 15 cycles using as a template RNA isolated from 90 h of MOF fermentation. Lane 1 icl1 primers for aceA encoding isocitrate lyase (see Table 1), lane 2 ms primers for aceB2 encoding malate synthase, lane 3 PCR control for DNA using ms primers. Source: Akopiants et al.

Heterologous expression of the S. coelicolor aceA in E. coli/pICL2. The aceA was cloned from pExIM1 into E. coli pBAD TOPO TA vector to provide pICL2. Data are presented as relative ICL activity in cell extracts generated from E. coli/pICL2 with (2) and without (1) arabinose induction. Source: Akopiants et al.
Cell extracts of S. cinnamonensis C730.1, L1, and L1/pExIM1 were assayed for both ICL and malate dehydrogenase activity. As shown in Fig. 1a, malate dehydrogenase catalyzes the subsequent step of the glyoxylate cycle after AceA (ICL activity) and AceB (MS activity). Whereas ICL activity was seen only for L1/pExIM1 (see above), clear and comparable levels of malate dehydrogenase activity were seen across all three strains for growth in both oil-based and TSB fermentation media.
Growth of S. cinnamonensis C730.1, L1, and L1/pEXIM1 on acetate as a sole carbon source
Streptomyces cinnamonensis C730.1, L1 mutant and L1/pExIM1 all grew on minimal medium with glucose as a sole carbon source (Fig. 7, plate 3), but could not grow in the absence of any carbon source (Fig. 7, plate 1). The C730.1 strain also had vigorous growth using acetate as a carbon source (Fig. 7, plate 2). In stark contrast neither the L1 nor L1/pExIM1 strains grew on acetate as a sole carbon source.

Growth of S. cinnamonensis L1 (A), L1/pExIM1 (B), and C730.1 (C) on minimal medium: plate 1 no carbon source, plate 2 sodium acetate as a sole carbon source, plate 3 glucose as a sole carbon source. Source: Akopiants et al.
Generation and analysis of UV mutants of S. cinnamonensis L1 and L1/pExIM1 able to grow on acetate as a sole carbon source
Spore suspensions of both S. cinnamonensis L1 and L1/pExIM1 were irradiated and then plated on minimal medium agar with acetate as a single carbon source. In the case of L1/pExIM1, 17 colonies, which grew on acetate as a sole carbon source were identified (Fig. 8, plate 1). Three of these, designated M1, M2, and M3, were selected for further analysis. Similarly 55 UV mutants of the L1 strain able to grow on acetate as a sole carbon source were obtained and three of these (MM1, MM2, and MM3) were selected for further study. We confirmed that all the isolated M mutants (Fig. 8, plate 2) could grow on acetate as a sole carbon source whereas the L1 strain could not. Nevertheless, these mutants and the L1 strain had the same phenotype when grown in an agar complex medium (Fig. 8, plate 3). Identical observations were made for the MM mutants (data not shown).

UV Mutants of S. cinnamonensis L1/pExIM1 able to grow on acetate as a sole carbon source. Plate 1 initial growth of irradiated spores on acetate, plate 2 seven isolated colonies from plate 1 (M1–M7) and the L1 strain (arrow) grown on the same minimal medium with acetate as a sole carbon source, plate 3 same as plate 2 but with a complex medium. Source: Akopiants et al.
The L1 mutant was obtained from the C730.1 strain by insertional inactivation of ccr with hyg (the hygromycin resistance gene) [12]. The M1-3 and MM1-3 mutants were all hygromycin resistant and PCR analysis of chromosomal DNA from each of these using the ccrc1 primer set confirmed the ccr insertional inactivation by hyg.
It has been shown previously that the majority of CCR activity in the C730.1 strain is attributable to CCR as this activity in cell extracts of the L1 mutant is reduced by at least 95% in the L1 strain relative to extracts of the C730.1 strain. Despite this decrease, residual CCR activity has been observed for cell extracts of the L1 mutant [12]. Intact incorporation of dual-labeled ethyl acetoacetate into the butyrate-derived positions of monensin in the L1 strain has also been reported (albeit reduced by at least 70% relative to that observed with the parental C730.1 strain). These two observations have led to speculation that another enoyl reductase in S. cinnamonensis may have CCR activity. Increased levels of such activity might thus permit the MM1-3 and M1-3 mutants of L1 and L1/pExIM1, respectively, to grow on acetate as a sole carbon source. To test this possibility we determined CCR activity in cell extracts of C730.1, L1, M1-3, and MM1-3. Clear CCR activity was observed only in cell extracts of C730.1. No CCR activity (limit of detection was 6% of activity observed for the C730.1 extracts) was observed in any of the other extracts.
We also carried out an incorporation study using dual labeled ethyl [3,4-13C2]acetoacetate, and observed the same low level intact labeling of methylmalonyl-CoA and ethylmalonyl-CoA derived positions of monensin A in both an M1 and MM3 mutant as well as the L1 strain. As the presence of CCR activity in C730.1 has been shown to be responsible for efficient labeling of both of these monensin positions from ethyl acetoacetate, this observation provides additional support ruling out an alternative enzyme providing CCR activity in either the M or the MM mutants.
The monensin titers of M1-3 and MM1-3 grown in oil-based fermentation medium were also evaluated in triplicate and all were shown to be within 5% of that previously reported for the L1 strain. It has been shown that under these fermentation conditions the L1 strain generates only 15% of the monensin titer observed for C730.1, as CCR is required for conversion of the acetyl-CoA to methylmalonyl-CoA via butyryl-CoA. Low monensin titers in both the M1-3 and MM1-3 mutants thus provide additional evidence against the presence of an additional enzyme providing CCR activity. Furthermore, whatever pathway allows these mutants to grow effectively on acetate as a sole carbon source, it would not appear to allow efficient conversion of acetyl-CoA to methylmalonyl-CoA in oil-based extended fermentation.
Finally, cell extracts of the MM1-3 mutants grown for 48 h in the final stages of an oil-based extended fermentation were examined for ICL activity (Fig. 4, bars 4–6). No detectable activity could be observed, whereas clear activity was seen for the L1/pExIM1 (Fig. 4, bar 1). The lack of ICL activity in the MM mutants suggests that despite their ability to grow on acetate as a sole carbon source, they do not appear to have an active glyoxylate cycle functioning in the oil-based extended fermentation medium.
Discussion
MS and ICLs are required for a functional glyoxylate pathway (Fig. 1a) [9]. Streptomyces cinnamonensis contains at least one gene (aceB) encoding a protein with high amino acid identity to AceB proteins with demonstrated MS activity. Streptomycetes, such as S. arenae, S. clavuligerus, S. coelicolor and S. griseus, have at least one aceB homologue (in most of these cases the gene has been expressed and the MS activity of the AceB protein confirmed) [6, 15]. RT-PCR analysis indicated that the S. cinnamonensis aceB was not expressed under the oil-based extended fermentation conditions. MS activity has been reported to be essential for growth of S. griseus and S. clavuligerus on acetate medium [2, 14], raising the possibility that under certain growth conditions the S. cinnamonensis aceB would be expressed. In the case of S. griseus and S. clavuligerus grown on acetate in liquid culture, the peak of MS activity was associated with the declining phase of cell growth, indicating that the enzyme was likely not required for initial growth. The function of the AceB and the corresponding MS activity in streptomycetes still needs to be determined.
Streptomycetes also have aceA genes, encoding AceA with ICL activity [20]. This has been shown clearly for S. clavuligerus [20] and in the current work for S. coelicolor. Despite the presence of both AceA and AceB, there is no evidence that streptomycetes use the glyoxylate cycle for growth on acetate. In fact the timing of ICL as well as MS activity in both S. griseus and S. clavuligerus grown on acetate in shake cultures suggests a late rather than early role. To the best of our knowledge, it is not known whether any streptomycete aceA and aceB is essential for liquid or solid phase growth on acetate.
In contrast, it has been shown that ccr is essential for solid phase growth of both S. collinus [4] and now S. cinnamonensis, on acetate. These observations are consistent with the proposal that the butyryl-CoA pathway (Fig. 1b), rather than the glyoxylate cycle is responsible for growth on acetate. It remains to be determined if ccr is similarly essential for solid-phase growth of other streptomycetes (S. griseus, S. clavuligerus, S. coelicolor).
The S. cinnamonensis strain L1/pExIM1 was constructed in order to determine if constitutive expression of the two critical glyoxylate cycle genes (aceA and aceB) would restore solid-phase growth on acetate, despite the block in the butyryl-CoA pathway. However, S. cinnamonensis L1/pExIM1 was unable grow on acetate, despite evidence demonstrating expression of both aceA and aceB, and the activity of the corresponding AceA (the activity of the aceB gene product has been demonstrated previously [15]). While it is possible that either these heterologous genes or other endogenous genes required for a functional glyoxylate cycle are not expressed under the solid phase growth conditions used (it is difficult to directly test this hypothesis as S. cinnamonensis L1/pExIM1 does not grow under the conditions used), it is likely that the issue may be more complex involving one or several other factors. For instance, it is known that channeling of isocitrate through the glyoxylate pathway is regulated via phosphorylation/dephosphorylation of isocitrate dehydrogenase (ID), the citric acid cycle enzyme, which competes with AceA for the isocitrate substrate [3]. When bacteria grow on acetate, phosphorylation of the ID by an ID kinase/phosphatase (whose expression is controlled by several transcriptional repressors and activators) leads to a dramatic decline in catalytic activity. A regulated decrease in ID activity alongside constitutive expression of aceA and aceB in S. cinnamonensis L1/pExIM1 might be necessary for a functional glyoxylate pathway and thus an ability to grow on acetate. Further studies are required to determine the conditions and regulatory mechanisms, if any, that will permit streptomycetes to grow on acetate using a glyoxylate cycle. The S. cinnamonensis L1 and L1/pExIM1 strains, unable to grow on acetate via the butyryl-CoA pathway, provide a good starting point for these investigations.
As a first step in this direction we used UV mutagenesis to obtain mutants of the L1 strain restored in their ability to grow on acetate, potentially via the glyoxylate cycle. Mutants able to grow on acetate occurred at essentially the same rate for both L1 and L1/pExIM1, suggesting that the pExIM1 plasmid does not have any significant effect, and that the heterologous expression of the S. coelicolor aceA and aceB may not be important. This observation, coupled with the absence of ICL activity in cell extracts of M1-3 mutants grown in the oil-based extended fermentation (where acetyl-CoA is the primary catabolite), provide some circumstantial evidence that these mutants may not be using the glyoxylate cycle for solid-phase growth on acetate. Analysis (evaluation of CCR activity, monensin production levels and monensin labeling by dual-labeled acetoacetate) in all of these mutants in liquid cultures indicated that the butyryl-CoA pathway (Fig. 1b) remained as ineffective as it was in the L1 mutant. Combined with genetic analysis, which confirmed that ccr was disrupted in these mutants, these observations provide circumstantial evidence that there is no butyryl-CoA pathway to permit solid-phase growth on acetate. Thus, it is possible that some other pathway or pathways allow such growth of these mutants. In this regard, it is noteworthy that previous investigations of S. cinnamonensis have provided evidence for a pathway or pathways, other than the CCR-dependent butyryl-CoA pathway, citric acid cycle, and glyoxylate cycle, linking acetyl-CoA/acetoacetyl-CoA to methylmalonyl-CoA/succinyl-CoA [22].
References
Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O’Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147
Chan M, Sim TS (1998) Malate synthase from Streptomyces clavuligerus NRRL3585: cloning, molecular characterization and its control by acetate. Microbiology 144(Pt 11):3229–3237
Cozzone AJ (1998) Regulation of acetate metabolism by protein phosphorylation in enteric bacteria. Annu Rev Microbiol 52:127–164
Han L, Reynolds KA (1997) A novel alternate anaplerotic pathway to the glyoxylate cycle in streptomycetes. J Bacteriol 179:5157–5164
Hopwood DA, Bibb MJ, Chater KF, Kieser T, Bruton CJ, Kieser HM, Lydiate DJ, Smith CP, Smith JM, Ward JM, Schrempf HS (1985) Genetic manipulation of streptomycetes, a laboratory manual. John Innes Institute, Norwich
Huttner S, Mecke D, Frohlich KU (1997) Gene cloning and sequencing, and enzyme purification of the malate synthase of Streptomyces arenae. Gene 188:239–246
Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol 21:526–531
Ivanovsky RN, Krasilnikova EN, Berg IA (1997) A proposed citramalate cycle for acetate assimilation in the purple non-sulfur bacterium Rhodospirillum rubrum. FEMS Microbiol Lett 153:399–404
Kornberg HL (1966) The role and control of the glyoxylate cycle in Escherichia coli. Biochem J 99:1–11
Korotkova N, Chistoserdova L, Kuksa V, Lidstrom ME (2002) Glyoxylate regeneration pathway in the methylotroph Methylobacterium extorquens AM1. J Bacteriol 184:1750–1758
Li C, Florova G, Konstatin A, Reynolds KA (2004) Crotonyl-coenzyme A reductase provides methylmalonyl-CoA precursors for monensin biosynthesis by Streptomyces cinnamonensis in an oil-based extended fermentation. Microbiology 150:3463–3472
Liu H, Reynolds KA (1999) Role of crotonyl coenzyme A reductase in determining the ratio of polyketides monensin A and monensin B produced by Streptomyces cinnamonensis. J Bacteriol 181:6806–6813
Loke P, Sim TS (2000) Molecular cloning, heterologous expression, and functional characterisation of a malate synthase gene from Streptomyces coelicolor A3(2). Can J Microbiol 46:764–769
Loke P, Wee J, Seah K, Sim TS (2001) PCR-mediated screening and cloning of a malate synthase gene from Streptomyces griseus NCIMB 9001. World J Microbiol Biotechnol 17:645–649
Loke P, Goh LL, Seng Soh B, Yeow P, Sim TS (2002) Purification and characterization of recombinant malate synthase enzymes from Streptomyces coelicolor A3(2) and S. clavuligerus NRRL3585. J Ind Microbiol Biotechnol 28:239–243
Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M (2001) Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci U S A 98:12215–12220
Park SJ, Cotter PA, Gunsalus RP (1995) Regulation of malate dehydrogenase (mdh) gene expression in Escherichia coli in response to oxygen, carbon, and heme availability. J Bacteriol 177:6652–6656
Reynolds KA, O’Hagan D, Gani D, Robinson JA (1988) Butyrate metabolism in streptomycetes. Characterization of a vicinal interchange rearrangement linking isobutyrate and butyrate in Streptomyces cinnamonensis. J Chem Soc Perkin Trans I 3195–3207
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Soh BS, Loke P, Sim TS (2001) Cloning, heterologous expression and purification of an isocitrate lyase from Streptomyces clavuligerus NRRL 3585. Biochim Biophys Acta 1522:112–117
Wallace KK, Bao Z, Dai H, Digate R, Schuler G, Speedie MK, Reynolds KA (1995) Purification of crotonyl CoA reductase from Streptomyces collinus and cloning, sequencing and expression of the corresponding gene in Escherichia coli. Eur J Biochem 233:954–962
Zhang W, Reynolds KA (2001) MeaA, a putative coenzyme B(12)-dependent mutase, provides methylmalonyl coenzyme A for monensin biosynthesis in Streptomyces cinnamonensis. J Bacteriol 183:2071–2080
Acknowledgments
We are grateful to Dr. H. Kieser for providing BAC 19F3 containing the S. coelicolor aceA and aceB. This work was supported by grants from the National Institute of Health (GM 50542) and Eli Lilly. We are grateful to Richard DeMaio and Vic Vinci at Eli Lilly for providing S. cinnamonensis C730.1 and for conditions and media for oil-based extended fermentations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Akopiants, K., Florova, G., Li, C. et al. Multiple pathways for acetate assimilation in Streptomyces cinnamonensis . J IND MICROBIOL BIOTECHNOL 33, 141–150 (2006). https://doi.org/10.1007/s10295-005-0029-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-005-0029-4