
Abstract
Using 11 microsatellite markers, we investigated the allelic variation and genetic structure of Cryptomeria japonica, across most of its natural distribution. The markers displayed high levels of polymorphism (average gene diversity=0.77, average number of alleles=24.0), in sharp contrast to the lower levels of polymorphism found in allozyme and cleaved amplified polymorphic sequence markers in previous studies. Little genetic differentiation was found among populations (FST=0.028, P<0.001), probably because the species is wind-pollinated and long-lived. No clear relationship between Nei’s genetic distances and geographical locations of the populations were found using the principal coordinate and unweighted pair-group method with arithmetic averaging analyses. The lack of such trends might be due partly to microsatellite homoplasy arising from mutation blurring the genealogical record. However, there was a trend towards high allelic diversity in five populations (Ashitaka, Ashiu, Oki-Island, Yakushima-Island-1 and -2), which are very close to, or in, refugial areas of the last glacial period as defined by Tsukada based on pollen analysis data and current climatic divisions. We postulate that these refugial populations might have been less affected by genetic drift than the other populations due to their relatively large size.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Populations in glacial refugia are generally expected to harbor higher levels of genetic diversity than similar populations in areas that have been colonized since the retreat of the glaciers (here termed “colonized populations”) because colonization often involves only a few individuals (Hewitt 1996). However, the ability of different kinds of genetic markers and analytical approaches to detect and resolve genetic diversity within or among populations differs markedly. At neutral loci, “bottlenecks” (transitory reductions in the effective population size) may theoretically have caused strong reductions in allelic richness, and a more limited decrease in gene diversity, since rare alleles are more readily affected by genetic drift than frequent alleles (Nei et al. 1975). Recent studies on European and North American tree species have shown that refugia and postglacial migration routes can be identified using DNA markers. Comps et al. (2001) reported evidence for a significant and steady decline in allelic richness during the postglacial recolonization of European beech using allozyme markers, but the expected reduction in gene diversity was not found. It has also been reported that allozyme variations in colonized populations decrease in other tree species (see, for instance, Tomaru et al. 1997).
Cryptomeria japonica is a wind-pollinated (anemophilous) species that produces abundant pollen every few years. Fossil pollen of this species has been analyzed in detail at many sites, allowing the identification of potential refugia and most probable routes of postglacial recolonization (Tsukada 1982). According to these studies, during the last glacial period, refugia of C. japonica occurred in several regions in Japan: the Izu Peninsula, along Wakasa Bay, Oki Island, Yaku Island, and probably from the Kii Peninsula to the southern part of Shikoku Island. In addition, there are two inferred recolonization pathways: one along the Sea of Japan side from the vicinity of Wakasa Bay, and the other along the Pacific side from the Izu Peninsula. Today’s natural forests are widely distributed in moist temperate regions from Aomori Prefecture (40°42′N) to Yakushima Island (30°15′N) (Hayashi 1960). However, the natural distribution is discontinuous and scattered in limited areas as a result of the extensive exploitation of this species during the past 1,000 years (Ohba 1993). Variations among natural forests of C. japonica in different regions have been investigated using morphological traits (needle length, needle curvature, and other features; Murai 1947) and diterpene components (Yasue et al. 1987). These studies have suggested that there are two main varieties: C. japonica ‘Omote-sugi’ (on the Pacific Ocean side of Japan) and C. japonica var. radicans ‘Ura-sugi’ (on the Sea of Japan side). This divergence is probably associated with the differences in their refugial origins in the last glacial period.
Genetic variation in natural populations of Cryptomeria japonica has been reported in studies based on allozymes (Tsumura and Ohba 1992, 1993; Tomaru et al. 1994) and cleaved amplified polymorphic sequence (CAPS) markers (Tsumura and Tomaru 1999). The studies cited found little genetic differentiation among populations, which is consistent with investigations of other conifers with a wide distribution. However, these studies did not find clear differences between the two main varieties, or between refugial and colonized populations, except between the Yakushima Island population and the other populations (Tsumura and Ohba 1993).
Like allozymes and CAPS markers, microsatellites have discrete and codominant alleles, but microsatellites have more alleles per locus and therefore allow higher resolution analysis. For instance, in a study by Chase et al. (1996), four polymorphic microsatellite loci provided greater resolution and sensitivity for the estimation of population genetic parameters than all of the allozyme markers analyzed previously in several detailed studies. Microsatellite variation might correlate directly with effective population size because populations with large effective sizes tend to retain high numbers of alleles originating from mutation, since the mutation rates of microsatellite loci are generally believed to be high compared to normal rates of point mutation (see, for instance, Jarne and Lagoda 1996). Microsatellite mutation rates of forest tree species such as conifers have not been investigated directly, and thus available information is limited to data on somatic mutation of Pinus strobus (Cloutier et al. 2003) and C. japonica (T. Takahashi et al. unpublished data). Both of these data sets suggest that microsatellite markers arising from somatic mutation tend to be stable. The influence of microsatellite mutation on the allelic composition of the markers used in the studies cited above may have been weak because of the longevity of C. japonica. If so, microsatellite variations of current natural populations might reflect the size and colonization history of past populations since the last glacial period.
Recently, microsatellite markers have been developed for Cryptomeria japonica in order to construct a genetic linkage map (Tani et al. 2003) and to investigate gene contamination in seed orchards (Moriguchi et al. 2003). In this paper, we discuss the allelic variation and population structure of C. japonica, as revealed by microsatellite markers, across most of its natural distribution. In addition we assess the availability and characteristics of microsatellite markers compared with the gene-coding markers (allozyme and CAPS markers; which have lower mutation rates than microsatellites) used in previous studies.
Materials and methods
Plant materials
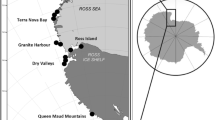
Mature needles were collected from 757 trees representing 29 populations. The locations of the sampled populations covered most of the natural distribution of Cryptomeria japonica (Fig. 1). All of these trees were from natural populations that have become established since the last glacial period, in forests that were designated candidates for an in situ gene-conservation program introduced by the Japanese Forestry agency, with the exception of the Tateyama, Sado-Island and Ashiu populations. We allowed at least 50 m between sampled trees to avoid sampling half-sibs. We also collected samples from trees as large in size as possible in old-growth forest to avoid sampling young trees that might have been influenced by artificial plantations. The needles were stored at −30°C before DNA extraction.

Natural distribution (Hayashi 1960) and locations of 29 surveyed natural populations of Cryptomeria japonica in Japan. The dotted line indicates the coastline ca. 18,000 years ago. Areas shaded in bold or with thin diagonal lines show refugia and probable refugia, respectively, at that time (Tsukada 1982)
According to suggestions by Tsukada (1982), based on pollen analysis and current climatic divisions, we divided the populations into three groups, defining populations in the vicinity of refugial areas of the last glacial period as refugia populations, populations in the vicinity of probable refugial areas in the last glacial period as probable refugia populations, and populations colonized after the last glacial as colonized populations (see Fig. 1, Table 1).
Laboratory analysis
Total DNA was extracted from these samples using a modified CTAB method (Tsumura et al. 1995). The genotypes of 11 microsatellite loci (Tani et al. 2003, 2004; Moriguchi et al. 2003) in each individual were determined. One of these 11 microsatellite loci (Cjs0201) was newly developed as a marker from a microsatellite-enriched library (Tani et al. 2004), and its sequence has been submitted to the DNA Data Bank of Japan (DDBJ). Of the 11 markers, 3 (Cjs0333, Cjs0520, and CS2169) were mapped to different linkage groups (Tani et al. 2003), but the locations of the other markers have not yet been mapped. PCR amplification was performed in 10-μl reaction volumes containing 3 ng genomic DNA, 1× PCR buffer (200 mM Tris-HCl, 500 mM KCl), 0.2 mM of each dNTP, 1.5 mM MgCl2, 0.2 μM of each primer (the forward primer of each pair labeled with dye), and 0.25 U Taq polymerase in a GeneAmp Model 9700 PCR System (PE Applied Biosystems, Foster City, Calif.). PCR conditions included a 5-min denaturing step at 94°C, followed by 30–35 cycles of 94°C for 30 s, the annealing temperature for 30 s, and 72°C for 30 s. The specific annealing temperatures for the primer pairs are shown in Table 1. Finally, there was an elongation step at 72°C for 5 min. The genotypes were determined using an ABI 3100 Genetic Analyzer and Genotyper software ver. 3.7 (PE Applied Biosystems), and confirmed manually.
Statistical analysis
To estimate within-population variation, we used four parameters calculated from the allele frequencies of all loci analyzed: the number of alleles (N a ), observed heterozygosity (H o ), Nei’s (1978) gene diversity (H e ), and allelic richness (for a minimum of six diploid individuals). Allelic richness was measured by the method of El Mousadik and Petit (1996), which is based on the rarefaction method of Hurlbert (1971). We also calculated the number of rare alleles (defined as alleles with a frequency <1% in the total population), and private alleles (unique to one population) per individual in each population. Nonparametric significance tests (the Kruskal-Wallis test with Bonferroni correction and the Mann-Whitney U test) were performed to examine differences in allelic diversity among defined groups.
Wright’s (1951) F-statistics were calculated to measure the deviation from Hardy-Weinberg equilibrium at each locus in each population. Fixation indices, FIS (used to estimate inbreeding within individuals in a population; the inbreeding coefficient), were calculated on the basis of Weir and Cockerham’s (1984) F values, with FSTAT version 2.9.3.2 (Goudet 2000). The significance of positive or negative values of FIS was calculated using 6,600 randomizations (the default value in FSTAT) for each locus and across loci for each population.
Genetic structure was assessed using two models: the infinite allele model (Kimura and Crow 1964), and the stepwise mutation model (SSM; Ohta and Kimura 1973; Kimura and Ohta 1978). Values of FST (Weir and Cockerham 1984), RST (Slatkin 1995), and hierarchical analysis of molecular variance (AMOVA) (Michalakis and Excoffier 1996) were calculated using the program ARLEQUIN, version 2.0 (Schneider et al. 1997), in which significance levels for the overall values were determined after 1,023 permutations. We also calculated RST values for all loci except Cjs0333, CS1364, CS1525 [for which the allelic frequencies did not follow SMM (Ohta and Kimura 1973)].
Associations between populations were analyzed by principal coordinate (PCO) analysis based on estimates of pairwise genetic similarities, calculated using the similarity coefficient of Nei (1978). The association was revealed by the first two PCO axes, which were found to be representative of higher-order axes. PCO allows underlying trends in the raw data to be visualized in a process involving data reduction by representation of the accessions in a space formed by newly constructed dimensions. Nei’s genetic distance (Nei 1978) was also calculated for each population pair, providing a basis for clustering populations by the unweighted pair-group method using arithmetic averages (UPGMA; Sokal and Seneath 1963).
Results
Genetic diversity at microsatellite loci
Levels of genetic diversity within each population were very high at all except two loci: CS1906 and CS2230. N a per locus ranged from 6 to 51, H e varied from 0.36 to 0.97, and H o from 0.21 to 0.95 (Table 2). The fixation indices (FIS) of CS1906 and CS2230 in each population showed significant positive deviations from the Hardy-Weinberg equilibrium. However, the mean values of FIS across populations for single loci varied from locus to locus (Table 2).
Genetic differentiation and structure of populations
To measure the degree of population differentiation, two theoretical models were used: the infinite alleles model (FST) and the stepwise mutation model (SMM; RST). The overall values of genetic differentiation for each model were similar, and each was significantly different from zero (P<0.001): FST=0.028, RST =0.032, RST (excluding Cjs0333, CS1364 and CS1525) =0.027.
The first two axes of the PCO analysis explained 39.3% of the total variation (Fig. 2). There seems to be slight clustering into two main varieties, Cryptomeria japonica ‘Omote-sugi’ (on the Pacific Ocean side) and C. japonica var. radicans ‘Ura-sugi’ (on the Sea of Japan side; Fig. 2). However, there was no clear relationship between genetic distances and geographical location of the populations from a dendrogram of genetic distance of these populations based on UPGMA (data not shown). PCO analysis also showed that Fuji populations were separated from the other populations (Fig. 2).

Genetic similarity among 29 populations, as revealed by principal coordinate (PCO) analysis. Open circles Cryptomeria japonica ‘Omote-sugi’ (on the Pacific Ocean side), closed circles Cryptomeria japonica var. radicans ‘Ura-sugi’ (on the Sea of Japan side of Japan)
Allelic diversity across natural populations
The mean values of the parameters used to estimate allelic diversity (i.e., allelic richness, and the numbers of rare and private alleles across populations) varied. In particular, there was a trend toward high allelic diversity in five populations (Ashitaka, Ashiu, Oki-Island, Yakushima-Island-1 and -2), which are very close to, or in, refugial areas of the last glacial period as defined by Tsukada (1982) based on pollen analysis data and current climate divisions. Three populations (Sanbe, Azouji and Oninome) in western Japan seem to have high allelic diversity. However, allelic diversity of the Shingu, Owase, Aki, Yanase populations, which are located in western Japan near probable refugial areas defined by Tsukada (1982), are relatively low.
Differences in allelic diversity amongst the three groups of Cryptomeria japonica populations (refugia, probable refugia and colonized populations, following proposals by Tsukada 1982), were found to be significant according to a Kruskal-Wallis test (P<0.05). More specifically, the allelic richness values, and the numbers of rare and private alleles between refugia and probable refugia populations differed significantly (P<0.05). In addition, although allelic richness values were not significantly different between refugia and colonized populations (P>0.05), there were significant differences in the numbers of rare and private alleles between refugia populations and colonized populations (P<0.05). There were no significant differences in mean gene diversity (H e ) between the three groups (Kruskal-Wallis test, P>0.05). Furthermore, when we compared the allelic diversity of C. japonica between eastern populations and western populations, there were no statistically significant differences in their values (Mann-Whitney U-test, P>0.05).
Discussion
Microsatellite markers in Cryptomeria japonica
In the present study, microsatellite markers displayed high levels of polymorphism (average gene diversity H e =0.770, average N a =24.0). This contrasts sharply with the low levels of polymorphism previously found in allozyme (average H e =0.189, average N a =2.3) and CAPS markers (average H e =0.281, average N a =1.9) in Cryptomeria japonica (Tomaru et al. 1994; Tsumura and Tomaru 1999). Our results indicate that microsatellite markers are much more informative than allozyme and CAPS markers in C. japonica, owing to their high polymorphism. The latter is attributable to their high mutation rates, which vary from 10−6 to 10−3 events per locus per generation (Hancock 1999), whereas mutation rates of allozyme loci are of the order of 10−6 events per locus per generation (see, for instance, Mukai and Cockerham 1977).
The average gene diversity over all loci in all populations exceeded the observed values, leading to positive mean values of FIS (Table 2). As a general explanation for these findings, a small excess of homozygotes may be due to non-random mating. Nevertheless, the mean values of FIS across populations found here varied from locus to locus (Table 2). Therefore, since inbreeding can be expected to affect all loci equally, there are likely to be additional causes of the deviations between observed and expected values, such as selection and subdivisions in the population structure. In addition, missamplification of microsatellite alleles, leading to the generation of “null alleles,” frequently occurs (Callen et al. 1993), and if null alleles occur at some loci, the gene diversity values may exceed the observed values.
Genetic structure of populations
Natural forests of Cryptomeria japonica show weak, but statistically significant, genetic differentiation among populations, as indicated by the average FST and RST values of 0.028 and 0.031, respectively, which are similar to GST values found in allozyme and CAPS marker studies (Tsumura and Ohba 1992, 1993; Tomaru et al. 1994; Tsumura and Tomaru 1999). The weak genetic differentiation between populations of C. japonica can be explained by the facts that C. japonica is a wind-pollinated, allogamous species with wind-dispersed seeds, and that long-lived woody species tend to have low levels of genetic variation among populations (Hamrick et al. 1992).
No clear geographical trends among populations were found in the PCO and UPGMA analyses. Although microsatellite loci often exhibit a large N a , this high diversity can also complicate the use of microsatellite data in evolutionary studies, such as the estimation of differentiation (Hedrick 1999) and genetic similarity between populations. One of the problems associated with microsatellite loci is their potential to demonstrate size homoplasy; a condition in which alleles that are identical in state (IIS) are not identical by descent (IBD) (Estoup and Cornuet 1999; Estoup et al. 2002). Homoplasy results from a mutation causing a proportion of alleles to either converge or diverge in state, blurring the genealogical record provided by IBD markers, and thus restricting our ability to infer the evolutionary processes that shape patterns of ancestry (Adams et al. 2004). We believe that the unclear results from PCO and UPGMA analyses might be due partly to microsatellite homoplasy. PCO analysis also showed that the Fuji population is separated from the other populations (Fig. 2). Although small, this population has unique alleles, presumably either through chance or because it has relatively high allelic diversity because of its location near to the refugial area of the last glacial period around Izu peninsula.
High allelic diversity in populations near refugial areas of the last glacial period
Values of the microsatellite allelic diversity parameters, i.e., allelic richness, and the numbers of rare and private alleles, were higher for the refugia populations than corresponding values for the other populations, but the expected H e values of the refugia populations were similar to those of the other populations. These findings are consistent with data related to European beech reported by Comps et al. (2001). In several studies, allozyme variation of colonized populations has been found to be lower than that of refugia populations (see, for instance, Tomaru et al. 1997). We postulated that refugia populations may have been less affected by genetic drift than the other populations due to their relatively large size, and that key characteristics of the species, especially its longevity (and thus the low number of generations since the last glacial period), may have helped maintain their relatively high levels of diversity.
In particular, there is a large area of natural Cryptomeria japonica forest on Yakushima Island, where natural forests of C. japonica grow at altitudes of 600–1,800 m, and some huge individuals are more than 1,000 years old. Furthermore, large C. japonica trees on Yakushima Island have been logged since the Edo-era, about 300 years ago, but logging was less intense there than on the mainland, because of the isolation of Yakushima Island from the mainland, and its steep mountains. Therefore, even now this natural forest covers about 8,000 ha at the center of the island and relatively high allelic diversity has been maintained within it.
Small populations that might be derived from main refugia of the last glacial period, such as the Ashitaka and Oki Island populations, still have high genetic diversity in terms of allelic richness, and rare and/or private allele frequencies. Today, natural forests of Cryptomeria japonica have declined in size because of the extensive exploitation of the species during the past 1,000 years (Ohba 1993), except on Yakushima Island. However, evidence from fossil pollen data (see, for instance, Tsukada 1982) and buried C. japonica forests, which have been found near Ashitaka Mountain (Takeda 1931) and Oki Island (Takahara et al. 2001), indicate that there were large natural forests of C. japonica nearby in the past. The current forest reserve areas of Fuji, Kawazu, Ashitaka and Oki Island, which were derived from main refugia, are very small (1.0, 0.8, 4.7 and 14.0 ha, respectively). However, those populations represent very important genetic resources because of their high genetic diversity and direct descent from refugia populations.
Allelic diversity of populations located near probable refugia (the Shingu, Owase, Aki, Yanase populations), which are located only on the Pacific Ocean side in western Japan, is low. From fossil pollen data, Takahara (1998) suggested that, in western Japan, especially on Sikoku Island, the expansion of natural forests of Cryptomeria japonica along the Pacific Ocean side might have been much slower than along the Japan Sea side because of the rapid expansion of evergreen broadleaf forest in the postglacial age. If there were refugia in those regions, their areas may have been small, and the chance of genetic drift greater than in the refugia populations.
We were able to show, using microsatellite markers, that the allelic diversity of refugia populations is higher than that of probable refugia and colonized populations. Today’s natural forest of Cryptomeria japonica is discontinuous, scattered in limited areas. On the other hand, C. japonica has been widely planted throughout Japan over an area of 4.5 million ha, accounting for 44% of all Japanese artificial forest. If the genetic diversity within populations of the artificial forests is the same as that of natural C. japonica forests, the genetic diversity of current natural forests of C. japonica should be maintained in the future. However, artificial forests may have less genetic diversity, because most of these forests in western Japan consist of limited numbers of clones that have been historically used as cultivar clones. Even in eastern Japan, the planted seedlings originate from seed orchards consisting of a limited number of mother trees. The genetic variation of natural C. japonica forests might be affected by pollen contamination from artificial forests in the future. Therefore, current natural forests of C. japonica are very valuable and important both for gene conservation and for future breeding programs. Information obtained from this study could be useful for conservation of the genetic resources of this species.
References
Adams RI, Brown KM, Hamilton MB (2004) The impact of microsatellite electromorph size homoplasy on multilocus population structure estimates in a tropical tree (Corythophora alta) and an anadromous fish (Morone saxatilis). Mol Ecol 13:2579–2588
Callen DF, Thompson AD, Shen Y, Phillips HA, Richards RI, Mulley JC, Sutherland GR (1993) Incidence and origin of “Null” alleles in the (AC)n microsatellite markers. Am J Hum Genet 52:922–927
Chase M, Kessell R, Bawa K (1996) Microsatellite markers for population and conservation genetics of tropical trees. Am J Bot 83:51–57
Cloutier D, Rioux D, Beaulieu J, Schoen DJ (2003) Somatic stability of microsatellite loci in Eastern white pine, Pinus strobus L. Heredity 90:247–252
Comps B, Gömöry D, Letouzey J, Thiebaut B, Petit RJ (2001) Diverging trends between hetrozygosity and allelic richness during postglacial colonization in the European beech. Genetics 157:389–397
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Estoup A, Cornuet JM (1999) Microsatellite evolution: inferences from population data. In: Goldstein DB, Schlötterer C (eds) Microsatellite: evolution and applications. Oxford University Press, Oxford, pp 49–65
Estoup A, Jarne P, Cornuet JM (2002) Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol Ecol 11:1591–1604
Goudet J (2000) FSTAT: a program to estimate and test gene diversities and fixation indices (version 2.9.1). Available at: http://www.unil.ch/izea/softwares/fstat.html
Hamrick JL, Godt MJW, Sherman-Broylers SL (1992) Factors influencing levels of genetic diversity in woody plant species. New For 6:95–124
Hancock JM (1999) Microsatellites and other simple sequences: genomic context and mutational mechanisms. In: Goldstein DB, Schlötterer C (eds) Microsatellite: evolution and applications. Oxford University Press, Oxford, pp 1–9
Hayashi Y (1960) Taxonomical and phytogeographical study of Japanese conifers (in Japanese). Norin-Shuppan, Tokyo
Hedrick PW (1999) Highly variable loci and their interpretation in evolution and conservation. Evolution 53:313–318
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276
Hurlbert SH (1971) The nonconcept of species diversity: a critique and alternative parameters. Ecology 52:577–586
Jarne P, Lagoda PJL (1996) Microsatellites, from molecules to populations and back. Trends Ecol Evol 11:424–429
Kimura M, Crow J (1964) The number of alleles that can be maintained in a finite population. Genetics 49:725–738
Kimura M, Ohta T (1978) Stepwise mutation model and distribution of allelic frequencies in a finite population. Proc Natl Acad Sci USA 75:2868–2872
Michalakis Y, Excoffier L (1996) A genetic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142:1061–1064
Moriguchi Y, Iwata H, Ujino-Ihara T, Yoshimura K, Taira H, Tsumura Y (2003) Development and characterization of microsatellite markers for Cryptomeria japonica D. Don. Theor Appl Genet 106:751–758
Mukai T, Cockerham CC (1977) Spontaneous mutation rate at enzyme loci in Drosophila melanogaster. Proc Natl Acad Sci USA 74:2514–2517
Murai S (1947) Major forestry tree species in the Tohoku region and their varietal problems (in Japanese). In: Kokudo Saiken Zourin Gijutsu Kouenshu, Aomori-rinyukai, Aomori
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Ohba K (1993) Clonal forestry with sugi (Cryptomeria japonica). In: Ahuja MR, Libby WJ (eds) Clonal forestry 2. Conservation and application. Springer, Berlin Heidelberg New York, pp 66–89
Ohta T, Kimura M(1973) The model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a genetic population. Genet Res 22:201–204
Schneider S, Kueffer JM, Roessli D, Excoffier L (1997) ARLEQUIN, version 2.0: software for population genetic data analysis. Genetic and Biometry Laboratory, University of Geneva, Switzerland. Available at: http://lgb.unige.ch/arlequin/
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Sokal RR, Seneath PHA (1963) Principals of numerical taxonomy. Freeman, San Francisco
Takahara H (1998) Sugi-rin no hensen. (in Japanese) In: Yasuda Y, Miyoushi N (eds) Nippon Rettou Syokusei-si. Asakura-Shoten, Tokyo, pp 207–223
Takahara H, Tanida K, Miyoshi N (2001) The full-glacial refuge of Cryptomeria japonica in the Oki Islands, western Japan. Jpn J Palynol 47:21–33
Takeda H (1931) Series of Japanese geography vol. 5 Mt. Fuji (in Japanese). Kaizo-sha, Tokyo
Tani N, Takahashi T, Iwata H, Mukai Y, Ujino-Ihara T, Matumoto A, Yoshimura K, Yoshimaru H, Murai M, Nagasaka K, Tsumura Y (2003) A consensus linkage map for sugi (Cryptomeria japonica) from two pedigrees, based on microsatellites and expressed tags. Genetics 165:1551–1568
Tani N, Takahashi T, Ujino-Ihara T, Iwata H, Yoshimura K, Tsumura Y (2004) Development and characteristics of microsatellite markers for sugi (Cryptomeria japonica D. Don) derived from microsatellite-enriched libraries. Ann Forensic Sci 61:569–575
Tomaru N, Tsumura Y, Ohba K (1994) Genetic variation and population differentiation in natural populations of Cryptomeria japonica. Plant Species Biol 9:191–199
Tomaru N, Mitsutsuji T, Takahashi M, Tsumura Y, Uchida K, Ohba K (1997) Genetic diversity in Fagus crenata (Japanese beech): influence of the distributional shift during the last-Quaternary. Heredity 78:241–251
Tsukada M (1982) Cryptomeria japonica: glacial refugia and late-glacial and postglacial migration. Ecology 63:1091–1105
Tsumura Y, Ohba K (1992) Allozyme variation of five natural populations of Cryptomeria japonica in western Japan. Jpn J Genet 67:299–308
Tsumura Y, Ohba K (1993) Genetic structure of geographical marginal populations of Cryptomeria japonica. Can J For Res 23:859–863
Tsumura Y, Tomaru N (1999) Genetic diversity of Cryptomeria japonica using co-dominant DNA markers based on sequenced-tagged sites. Theor Appl Genet 98:396–404
Tsumura Y, Yoshimura K, Tomaru N, Ohba K (1995) Molecular phylogeny of conifers using RFLP analysis of PCR-amplified specific chloroplast genes. Theor Appl Genet 91:1222–1236
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wright S (1951) The genetic structure of populations. Ann Eugen 15:323–354
Yasue M, Ogiyama K, Suto S, Tsukahara H, Miyahara F, Ohba K (1987) Geographical differentiation of natural Cryptomeria stands analyzed by diterpene hydrocarbon constituents of individual trees. J Jpn For Soc 69:152–156
Acknowledgements
The authors are grateful to H. Iwata, S. Ueno, S. Kanetani and T. Kado for valuable suggestions and technical support. We also thank M. Sakimoto, K. Hirayama, A. Matsumoto, T. Ihara, K. Yoshimura, K. Nagasaka, and Y. Moriguchi for their help in sample collection. The study was partly supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (no. 16380112) and a grant for Research on Conservation of Endemic Tree Species and Genetic Diversity in the Ecosystem of the Yakushima Islands from the Ministry of Environment, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takahashi, T., Tani, N., Taira, H. et al. Microsatellite markers reveal high allelic variation in natural populations of Cryptomeria japonica near refugial areas of the last glacial period. J Plant Res 118, 83–90 (2005). https://doi.org/10.1007/s10265-005-0198-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-005-0198-2