
Abstract
The reprogramming of cellular metabolism is a hallmark of tumorigenesis. However, the prognostic value of metabolism-related genes in colon cancer remains unclear. This study aimed to identify a metabolic gene signature to categorize colon cancer patients into high- and low-risk groups and predict prognosis. Samples from the Gene Expression Omnibus database were used as the training cohort, while samples from The Cancer Genome Atlas database were used as the validation cohort. A metabolic gene signature was established to investigate a robust risk stratification for colon cancer. Subsequently, a prognostic nomogram was established combining the metabolism-related risk score and clinicopathological characteristics of patients. A total of 351 differentially expressed metabolism-related genes were identified in colon cancer. After univariate analysis and least absolute shrinkage and selection operator-penalized regression analysis, an eight-gene metabolic signature (MTR, NANS, HADH, IMPA2, AGPAT1, GGT5, CYP2J2, and ASL) was identified to classify patients into high- and low-risk groups. High-risk patients had significantly shorter overall survival than low-risk patients in both the training and validation cohorts. A high-risk score was positively correlated with proximal colon cancer (P = 0.012), BRAF mutation (P = 0.049), and advanced stage (P = 0.027). We established a prognostic nomogram based on metabolism-related gene risk score and clinicopathologic factors. The areas under the curve and calibration curves indicated that the established nomogram showed a good accuracy of prediction. We have established a novel metabolic gene signature that could predict overall survival in colon cancer patients and serve as a biomarker for colon cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is one of the most common intestinal malignancies worldwide. It has the third highest incidence among cancers and remains the second most common cause of cancer mortality [1]. Due to new understanding of genetic factors in cancer, the introduction of screening tests, and the improvement of treatment, the morbidity and mortality of CRC have declined [2]. However, CRC is a highly heterogeneous disease [3]. The postoperative survival rates of CRC patients with different stages vary widely. For example, the 5-year survival rate for patients with stage IV CRC is < 10%, while that for patients with stage I CRC is > 90% [4]. Therefore, it is very crucial to identify effective methods to improve strategies for CRC diagnosis and prognosis.
Evidence suggests that tumorigenesis involves various changes in cellular metabolism. Rapidly growing cancer cells produce ATP at a high rate of glycolysis, independent of the availability of oxygen, in a process known as the Warburg effect [5]. This change in the metabolic pathway is a hallmark of cancer because it helps the absorption of large amounts of nutrients into cellular building blocks (nucleotides, amino acids, and lipids), causing excess production of the antioxidant glutathione and thereby producing new cells [6, 7]. In most cases, metabolic changes are driven by oncogene-directed metabolic reprogramming [8], which seems to be a common feature of highly malignant tumors [9], independent of their carcinogenic origin [10]. Therefore, it is necessary to study the characterization of metabolic pathways to understand how tumors regulate metabolic phenotypes. Recent papers describe several abnormally expressed metabolic genes in CRC that are associated with survival [11, 12]. However, the expression patterns and mechanisms of these genes that mediate metabolic reprogramming are unclear.
This study aimed to identify differentially expressed metabolic genes and establish a useful signature from these genes to predict clinical outcomes and guide treatment decisions in patients with colon cancer. Particularly, we established and validated an eight-gene metabolic signature based on data from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases. Additionally, associations between the risk score and clinicopathological features were evaluated. The signature predicted not only survival but also aggressive clinical outcomes in colon cancer patients.
Materials and methods
Data source
We downloaded the GSE39582 gene expression profile from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and RNA sequencing (RNA-seq) data from the TCGA database (https://portal.gdc.cancer.gov/). The GEO dataset included 566 colon cancer samples and 19 normal renal tissue samples, while the TCGA dataset included 473 colon cancer samples and 41 normal colon tissue samples. Seventy-one patients in the GEO cohort were excluded due to unknown tumor stage (n = 5), KRAS mutation status (n = 26), and BRAF mutation status (n = 40). Among the 437 colon cancer patients in the TCGA cohort, 34 were excluded as the number of survival months (n = 23) and tumor stage (n = 11) was unknown. Overall, 495 samples from the GEO dataset were used as the training cohort, and 403 samples from the TCGA dataset were used as the validation cohort. This study did not require ethics approval as all data were downloaded from the GEO and TCGA, which are publicly available databases.
Differentially expressed analyses
We used the “limma” package to test all the data to identify metabolic genes that were differentially expressed by an absolute log fold change (|log FC|) of > 1 and an adjusted P value of < 0.05 in the tumor samples compared with the normal samples.
Functional analysis of metabolic genes
The functions of the metabolic genes were analyzed using the Gene Ontology (GO) tool and the Kyoto Encyclopedia of Genes and Genomes (KEGG) and using the “clusterProfiler” package (Bioconductor) [13]. A P-value of < 0.05 was considered significant.
Development and validation of the metabolic gene signature
On univariate Cox regression analysis, genes with P < 0.01 were defined as candidate genes for further least absolute shrinkage and selection operator (LASSO)-penalized Cox regression analysis. The regression coefficients of each related gene were reserved for developing the following formula for the risk score: risk score = (mRNA1 coefficient × mRNA1 expression) + (mRNA2 coefficient × mRNA2 expression) + ··· + (mRNAn coefficient × mRNAn expression). Based on the median risk score, we divided the colon cancer patients into high- and low-risk groups. Differences in overall survival (OS) were compared and analyzed using the Kaplan–Meier method and log-rank test. The predictive ability of the metabolic gene signature was assessed based on the time-dependent receiver operating characteristic (ROC) curve and was validated using data from the TCGA database. The risk score for each patient was calculated with the same formula as the training cohort.
Construction of a prognostic nomogram
To explore whether the metabolic gene signature was independent of other clinicopathological parameters (including age, gender, tumor location, KRAS mutation, BRAF mutation, and tumor stage), univariate and multivariate Cox regression analyses were conducted. Based on the results of the multivariate Cox regression analysis, we constructed a nomogram for predicting 3-year and 5-year OS through the use of the “rms” package. Discrimination and calibration were measured to determine the validity of the nomogram. Discrimination was evaluated using the area under the ROC curve (AUC) [14]. The AUC ranged from 0.5 to 1.0, with 0.5 indicating a totally random outcome and 1.0 indicating perfect discrimination. We compared the nomogram and the American Joint Committee on Cancer (AJCC) TNM staging system based on the AUC. A calibration map was generated by comparing the nomogram’s prediction probability and the observation for the 3-year and 5-year OS rates.
Statistical analyses
Continuous variables are presented as means and standard deviations, as appropriate. The χ2 test and t-tests were used to compare differences between two groups. The predictive performances of the metabolic gene signature and nomogram were assessed based on the AUC using the “survivalROC” package in R. All statistical analyses were performed with SPSS (version 23.0; Chicago, IL, USA) and R software (version 3.5.3). A P-value of < 0.05 was defined to be statistically significant.
Results
Patient characteristics
Table 1 presents the clinical characteristics of colon cancer patients. A total of 898 eligible patients with colon cancer were included in the study—495 patients were in the training cohort and 403 were in the validation cohort. In both cohorts, most of the patients were male and aged > 60 years. In the training cohort, the most common tumor site was the distal colon (59.8%), and most patients did not have KRAS (60.6%) or BRAF mutation (90.1). In the validation cohort, data on KRAS and BRAF mutation status were missing in > 90% of patients. In both cohorts, the most common stage was stage II, followed by stage III and stage I or IV.
Identification of differentially expressed metabolic genes
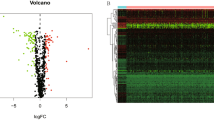
After intersecting with metabolic genes in the TCGA database, a total 863 metabolic genes remained. In the expression profiles of colon cancer compared with the normal colon tissues in the GSE39582 dataset, we identified 351 differentially expressed metabolic genes with |log FC| > 2 and P < 0.01. Of these genes, 176 were downregulated and 175 were upregulated (Fig. 1).

Heatmap of differentially expressed metabolic genes in the Gene Expression Omnibus (GEO) cohort
Functional enrichment analysis
To uncover the biological processes of the 351 differentially expressed metabolic genes in colon cancer patients, we performed a GO annotation. A total of 714 GO terms (including 547 biological processes, 30 cellular components, and 136 molecular functions) were enriched (Fig. 2a). The GO biological processes mainly comprised small-molecule catabolic processes; cellular components, the mitochondrial matrix; and molecular functions, cofactor binding. Furthermore, analysis of the KEGG pathways of these metabolic genes showed that 63 KEGG pathways were enriched, mainly including purine metabolism, carbon metabolism, glutathione metabolism, and drug metabolism (Fig. 2b).

Enrichment of top 10 GO terms (a) and KEGG pathways (b) of differentially expressed metabolic genes. The node color changes gradually from red to blue in ascending order according to the adjusted P-values. The node size represents the number of counts. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes (color figure online)
Establishment of the metabolic gene signature in the GEO cohort
To identify which genes were related to OS, the 351 differentially expressed metabolic genes were initially subjected to univariate Cox proportional hazards regression analysis in the GEO cohort. The results showed that 31 metabolic genes were significantly associated with OS (P < 0.01). Finally, eight genes were identified by the LASSO-penalized Cox regression model and were used to establish the metabolic gene signature; these were MTR, NANS, HADH, IMPA2, AGPAT1, GGT5, CYP2J2, and ASL (Fig. 3). The risk score formula was (−0.0454 × MTR expression) + (−0.0191 × NANS expression) + (−0.0131 × HADH expression) + (−0.0134 × IMPA2 expression) + (0.0214 × AGPAT1 expression) + (0.0036 × GGT5 expression) + (−0.0117 × CYP2J2 expression) + (−0.0241 × ASL expression). The risk scores of the 495 patients were calculated according to the formula. All patients were assigned to high- (n = 248) and low-risk (n = 248) groups based on median risk scores. The Kaplan–Meier analysis, risk score analysis, and time-dependent ROC analysis in the GEO cohort are shown in Fig. 4a. According to the Kaplan–Meier log-rank test, there were significant differences in OS between the two groups (P < 0.007; Fig. 4a). The AUC value for OS was 0.754, indicating a good ability of the signature to predict survival risk in colon cancer patients (Fig. 4a).

Expression levels of eight differentially expressed metabolic genes in colon cancer

Risk score analysis, Kaplan–Meier analysis, and time-dependent ROC curve analysis of the eight-gene metabolic signature in the GEO cohort (a) and TCGA cohort (b). ROC, receiver operating characteristic; GEO, Gene Expression Omnibus; TCGA, The Cancer Genome Atlas
Validation of the metabolic gene signature in the TCGA cohort
To validate the predictive ability of the metabolic gene signature, risk scores were calculated with the same formula for the TCGA cohort. Consistent with the results in the GEO cohort, patients in the high-risk group showed significantly worse OS (P = 0.032) (Fig. 4b). The risk score analysis, Kaplan–Meier analysis, and time-dependent ROC analysis in the GEO cohort are shown in Fig. 4b. For this validation cohort, the AUC for OS was 0.663. Taking both the training and validation cohorts together, we found that the metabolic gene signature was capable of predicting OS in colon cancer.
Correlation between the risk score and clinicopathological features
An analysis was performed to evaluate the association between the risk score and patient age, sex, tumor location, KRAS mutation, BRAF mutation, and tumor stage. The results (Table 2) demonstrated that a high-risk score was positively correlated with proximal colon cancer (P = 0.012), BRAF mutation (P = 0.049), and advanced stage (P = 0.027).
Construction of the nomogram in the GEO cohort
As shown in Table 3, univariate and multivariate analyses were used to find independent prognostic factors for OS in colon cancer patients. We found that patient age, sex, KRAS mutation, tumor stage, and risk score were risk factors for colon cancer (P < 0.05). These factors were selected to construct the gene-clinical nomogram of OS in colon cancer patients (Fig. 5). In the time-dependent ROC curve, the nomogram could effectively predict the 3- and 5-year survival rates (AUC, 0.768 and 0.745, respectively; Fig. 6). In addition, the predictive ability of our nomogram was significantly superior to that of the TNM staging system (Fig. 6a). The calibration curves of 3- and 5-year survival probability exhibited good agreement with the actual observed values (Fig. 6b). Moreover, we evaluated the prognosis of patients stratified by age and tumor stage in both the training and validation cohorts. The Kaplan–Meier curve analysis also indicated that the OS of patients with stage IV cancer was significantly shorter than that of patients with other stages in the training and validation cohorts (Fig. 7a, b). However, there were no significant differences in OS between the two groups in the training and validation cohorts (< 50 and ≥ 50) (Fig. 7c, d).

Nomogram for predicting 3- and 5-year overall survival in colon cancer patients

Comparison of a the AUCs of the nomogram and the American Joint Committee on Cancer (AJCC) TNM staging system and b calibration curves of the nomogram for predicting 3-year and 5-year survival in colon cancer patients

Kaplan–Meier analysis of overall survival stratified by tumor stage (a, b) and age (c, d) in the training and validation cohorts
Discussion
According to the American Cancer Society, there were approximately 1.8 million new cases of CRC and 881,000 deaths from CRC in 2018, which accounted for nearly 10% of new cancer cases and deaths in the same year worldwide [1]. With continuing progress in CRC screening and diagnosis in developing countries, the number of new CRC cases worldwide is predicted to increase to 2.5 million in 2035 [15]. A worrying rise in patients presenting with CRC at the age of < 50 years has also been observed [16,17,18]. Typically, the ideal treatment for CRC is surgical intervention, but 25% of CRC patients are diagnosed at an advanced stage with metastases, which result in difficulties in curative surgical control [19]. Patients who cannot undergo surgery mainly receive radiation therapy, chemotherapy, or immunotherapy. However, these conventional treatments are associated with adverse effects, such as resistance to chemotherapy, systemic toxicity, and cancer recurrence [20, 21]. These challenges call for an urgent need to find more effective methods to improve strategies for CRC diagnosis and prognosis.
Metabolic reprogramming is considered a hallmark of cancer because it is the most common physiological change in cancer cells [22]. Colon cancer undergoes severe metabolic reprogramming during its transformation, and changes in metabolic processes can be recognized by system biology tools [23]. Therefore, understanding changes in metabolite levels associated with colon cancer will help in the search for accurate and clinically useful biomarkers and therapeutic targets. Comprehensive analysis of metabolomics and transcriptomics data is an advanced method for finding reliable metabolic biomarkers. In recent years, several metabolic gene models have been established for various types of cancer, such as hepatocellular carcinoma [24], thyroid cancer [25], and ovarian cancer [26]. However, to our knowledge, no metabolic gene signature has been established for colon cancer. Therefore, this study attempted to identify differentially expressed metabolic genes and establish a useful metabolic gene signature to predict clinical outcomes for patients with colon cancer.
In this study, we identified dysregulated metabolic genes in colon cancer using GEO and TCGA data. A novel eight-gene metabolic signature (MTR, NANS, HADH, IMPA2, AGPAT1, GGT5, CYP2J2, and ASL) was established in the GEO cohort, and the signature demonstrated a robust predictive ability in the TCGA cohort. We also found that a high-risk score was positively correlated with proximal colon cancer, BRAF mutation, and advanced tumor stage in colon cancer. This finding further supports the robustness of the prognostic value of the signature. Furthermore, a prognostic nomogram was established based on the risk score and four clinicopathological factors. The prognostic accuracy of the model was confirmed by the ROC curve and the corresponding calibration curve. The predictive ability of our nomogram was significantly superior to that of the TNM staging system. These results indicate that the signature not only serves as a biomarker for colon cancer, independent of clinicopathological features, but also predicts clinical outcomes in colon cancer patients.
We identified a total of 351 differentially expressed metabolic genes. Analysis of the KEGG pathways indicated that these genes were mainly enriched in purine, carbon, glutathione, and drug metabolism. Most enrichment pathways are related to metabolism, and the significant genes of enrichment pathways are mainly involved in purine metabolism, which is an important pathway that provides nucleotides for aggressive DNA synthesis in cancer cells [27]. Purine is an essential component of nucleotides in cell proliferation, so purines and enzymes for de novo purine biosynthetic pathways are enhanced in tumor cells [28, 29]. Accumulating evidence suggests that disturbed purine metabolism was found in various types of cancer, including CRC [30,31,32]. In CRC tissue, purine metabolism and the rescue pathway activity of purine nucleotides are accelerated [33]. Vannoni et al. [34] found that enzyme activity in colorectal tumor tissue is significantly higher than that in normal tissue to cope with the accelerated purine metabolism. Other metabolic pathways were also reported to be linked to tumorigenesis in colon cancer [35, 36]. Thus, these differentially expressed metabolic genes influence the occurrence and development of colon cancer through their mediated metabolic pathways.
Most of the eight genes of the signature have been reported in various types of cancer, including CRC [37,38,39,40]. AGPAT1 (1-acylglycerol 3-phosphate O-acyltransferase 1) is a lipid metabolism-related gene whose overexpression has been significantly associated with CRC prognosis, suggesting that AGPAT1 may serve as a prognostic indicator of CRC [38]. In our study, we also demonstrated that NRG1 was downregulated in colon cancer and associated with worse OS. Meanwhile, Zara-Lopes et al. [39] found that methionine synthase (MTR) 2756A > G polymorphism was linked to tumor extent and aggressiveness in thyroid cancer. Shen et al. [40] indicated that the expression of HADH (3-hydroxyacyl-CoA dehydrogenase) was significantly downregulated in gastric cancer tissues. HADH silencing can significantly promote migration and invasion of gastric cancer cells by activating the Akt signaling pathway.
This study has some limitations. First, there were no data on tumor location in the TCGA cohort, and data on KRAS mutation status were missing for > 90% of the TCGA cohort. Second, we were unable to evaluate the predictive accuracy of the nomogram by external validation. Lastly, although the signature of the eight metabolic genes showed favorable predictive ability in colon cancer, the exact mechanism of each gene remains unclear. Therefore, external data from large independent cohorts are necessary to validate the characteristics and prognostic nomograms of the eight metabolic genes. Moreover, further functional experiments in colon cancer are needed to explore the elusive mechanisms of aberrant metabolic pathways caused by the eight metabolic genes.
Conclusions
We successfully established a novel metabolic gene signature in colon cancer. This signature was not only able to predict OS in colon cancer patients but was also significantly associated with poor clinical outcomes.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544–73. https://doi.org/10.1002/cncr.24760.
Ullmann P, Nurmik M, Begaj R, Haan S, Letellier E. Hypoxia- and MicroRNA-Induced Metabolic Reprogramming of Tumor-Initiating Cells. Cells. 2019. https://doi.org/10.3390/cells8060528.
O’Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst. 2004;96(19):1420–5. https://doi.org/10.1093/jnci/djh275.
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14. https://doi.org/10.1126/science.123.3191.309.
Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71(22):6921–5. https://doi.org/10.1158/0008-5472.Can-11-1457.
Gao P, Tchernyshyov I, Chang TC, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–5. https://doi.org/10.1038/nature07823.
Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. https://doi.org/10.1016/j.ccr.2012.02.014.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Bustamante E, Morris HP, Pedersen PL. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J Biol Chem. 1981;256(16):8699–704.
Li S, Xie L, Du M, et al. Association study of genetic variants in estrogen metabolic pathway genes and colorectal cancer risk and survival. Arch Toxicol. 2018;92(6):1991–9. https://doi.org/10.1007/s00204-018-2195-y.
Ose J, Botma A, Balavarca Y, et al. Pathway analysis of genetic variants in folate-mediated one-carbon metabolism-related genes and survival in a prospectively followed cohort of colorectal cancer patients. Cancer Med. 2018. https://doi.org/10.1002/cam4.1407.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. https://doi.org/10.1089/omi.2011.0118.
Wolbers M, Koller MT, Witteman JC, Steyerberg EW. Prognostic models with competing risks: methods and application to coronary risk prediction. Epidemiology. 2009;20(4):555–61. https://doi.org/10.1097/EDE.0b013e3181a39056.
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet (London, England). 2019;394(10207):1467–80. https://doi.org/10.1016/S0140-6736(19)32319-0.
Wolf AMD, Fontham ETH, Church TR, et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J Clin. 2018;68(4):250–81. https://doi.org/10.3322/caac.21457.
Kasi PM, Shahjehan F, Cochuyt JJ, Li Z, Colibaseanu DT, Merchea A. Rising proportion of young individuals with rectal and colon cancer. Clin Colorectal Cancer. 2019;18(1):e87–95. https://doi.org/10.1016/j.clcc.2018.10.002.
Lasabová Z, Kalman M, Holubeková V, et al. Mutation analysis of POLE gene in patients with early-onset colorectal cancer revealed a rare silent variant within the endonuclease domain with potential effect on splicing. Clin Exp Med. 2019;19(3):393–400. https://doi.org/10.1007/s10238-019-00558-7.
Vatandoust S, Price TJ, Karapetis CS. Colorectal cancer: metastases to a single organ. World J Gastroenterol. 2015;21(41):11767–76. https://doi.org/10.3748/wjg.v21.i41.11767.
Singh B, Mitragotri S. Harnessing cells to deliver nanoparticle drugs to treat cancer. Biotechnol Adv. 2019. https://doi.org/10.1016/j.biotechadv.2019.01.006.
Zhang Y, Yuan J, Zhang HY, et al. Natural resistance to apoptosis correlates with resistance to chemotherapy in colorectal cancer cells. Clin Exp Med. 2012. https://doi.org/10.1007/s10238-011-0146-5.
Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res. 2017;7(5):1016–36.
Zhang C, Aldrees M, Arif M, Li X, Mardinoglu A, Aziz MA. Elucidating the reprograming of colorectal cancer metabolism using genome-scale metabolic modeling. Front Oncol. 2019;9:681. https://doi.org/10.3389/fonc.2019.00681.
Liu GM, Xie WX, Zhang CY, Xu JW. Identification of a four-gene metabolic signature predicting overall survival for hepatocellular carcinoma. J Cell Physiol. 2019. https://doi.org/10.1002/jcp.29081.
Ma B, Jiang H, Wen D, et al. Transcriptome analyses identify a metabolic gene signature indicative of dedifferentiation of papillary thyroid cancer. J Clin Endocrinol Metab. 2019;104(9):3713–25. https://doi.org/10.1210/jc.2018-02686.
Motamedian E, Taheri E, Bagheri F. Proliferation inhibition of cisplatin-resistant ovarian cancer cells using drugs screened by integrating a metabolic model and transcriptomic data. Cell Prolif. 2017. https://doi.org/10.1111/cpr.12370.
Kuroda K, Fukuda T, Isogai H, Okumura K, Krstic-Demonacos M, Isogai E. Antimicrobial peptide FF/CAP18 induces apoptotic cell death in HCT116 colon cancer cells via changes in the metabolic profile. Int J Oncol. 2015;46(4):1516–26. https://doi.org/10.3892/ijo.2015.2887.
Bahreyni A, Samani SS, Rahmani F, et al. Role of adenosine signaling in the pathogenesis of breast cancer. J Cell Physiol. 2018;233(3):1836–43. https://doi.org/10.1002/jcp.25944.
Goswami MT, Chen G, Chakravarthi BV, et al. Role and regulation of coordinately expressed de novo purine biosynthetic enzymes PPAT and PAICS in lung cancer. Oncotarget. 2015;6(27):23445–61. https://doi.org/10.18632/oncotarget.4352.
Barfeld SJ, Fazli L, Persson M, et al. Myc-dependent purine biosynthesis affects nucleolar stress and therapy response in prostate cancer. Oncotarget. 2015;6(14):12587–602. https://doi.org/10.18632/oncotarget.3494.
Wang X, Yang K, Xie Q, et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat Neurosci. 2017;20(5):661–73. https://doi.org/10.1038/nn.4537.
Yin J, Ren W, Huang X, Deng J, Li T, Yin Y. Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front Immunol. 2018;9:1697. https://doi.org/10.3389/fimmu.2018.01697.
Eroglu A, Canbolat O, Demirci S, Kocaoglu H, Eryavuz Y, Akgül H. Activities of adenosine deaminase and 5′-nucleotidase in cancerous and noncancerous human colorectal tissues. Med Oncol. 2000;17(4):319–24. https://doi.org/10.1007/bf02782198.
Vannoni D, Di Pietro MC, Rosi F, et al. Metabolism of adenosine in human colorectal tumour. Nucleosides Nucleotides Nucleic Acids. 2004;23(8–9):1455–7. https://doi.org/10.1081/ncn-200027676.
Asante I, Chui D, Pei H, et al. Alterations in folate-dependent one-carbon metabolism as colon cell transition from normal to cancerous. J Nutr Biochem. 2019;69:1–9. https://doi.org/10.1016/j.jnutbio.2019.02.008.
Lopes-Ramos CM, Kuijjer ML, Ogino S, et al. Gene regulatory network analysis identifies sex-linked differences in colon cancer drug metabolism. Cancer Res. 2018;78(19):5538–47. https://doi.org/10.1158/0008-5472.Can-18-0454.
Voloshanenko O, Schwartz U, Kranz D, et al. β-catenin-independent regulation of Wnt target genes by RoR2 and ATF2/ATF4 in colon cancer cells. Sci Rep. 2018;8(1):3178. https://doi.org/10.1038/s41598-018-20641-5.
Fernández LP, Ramos-Ruiz R, Herranz J, et al. The transcriptional and mutational landscapes of lipid metabolism-related genes in colon cancer. Oncotarget. 2018;9(5):5919–30. https://doi.org/10.18632/oncotarget.23592.
Zara-Lopes T, Galbiatti-Dias ALS, Castanhole-Nunes MMU, et al. Polymorphisms in, and genes involved in folate metabolism and thyroid cancer: a case-control study. Arch Med Sci. 2019;15(2):522–30. https://doi.org/10.5114/aoms.2018.73091.
Shen C, Song YH, Xie Y, et al. Downregulation of HADH promotes gastric cancer progression via Akt signaling pathway. Oncotarget. 2017;8(44):76279–89. https://doi.org/10.18632/oncotarget.19348.
Funding
None.
Author information
Authors and Affiliations
Contributions
JR and JF designed the experiment. JR, JF, WS, and CTW undertook the data acquisition. JR, JF, WS, and YHG were involved in the interpretation of data. JR and JF analyzed and visualized the data. All authors drafted and revised the manuscript. The final manuscript was read and approved by all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ren, J., Feng, J., Song, W. et al. Development and validation of a metabolic gene signature for predicting overall survival in patients with colon cancer. Clin Exp Med 20, 535–544 (2020). https://doi.org/10.1007/s10238-020-00652-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-020-00652-1