
Abstract
Background
The importance of metabolism-related alterations in the development of gastric cancer (GC) is increasingly recognized. The present study aimed to identify metabolism-related genes to facilitate prognosis of GC patients.
Methods
Gene expression datasets and clinical information of GC patients were downloaded from TCGA and GEO databases. We scored the enrichment of human metabolism-related pathways (n = 86) in GC samples by GSV, constructed prognostic risk models using LASSO algorithm and multivariate Cox regression analysis, combined with clinical information to construct a nomogram, and finally cis score algorithm to analyze the abundance of immune-related cells in different subtypes. We used Weka software to screen for prognosis-related marker genes and finally validated the expression of the selected genes in clinical cancer patient tissues.
Results
We identified that two GC metabolism-related signatures were strongly associated with OS and the levels of immune cell infiltration. Moreover, a survival prediction model for GC was established based on six GC metabolism-related genes. Time-dependent ROC analysis showed good stability of the risk prediction scoring model. The model was successfully validated in an independent ACRG cohort, and the expression trends of key genes were also verified in the GC tissues of patients. DLX1, LTBP2, FGFR1 and MMP2 were highly expressed in the cluster with poorer prognosis while SLC13A2 and SLCO1B3 were highly expressed in the cluster with better prognosis.
Conclusions
We identified a risk predictive score model based on six metabolism-related genes related to survival, which may serve as prognostic indicators and potential therapeutic targets for GC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastric cancer (GC) is the fifth-most common cancer globally and the fourth-leading cause of cancer deaths. It accounts for 1,080,800 cases annually, with the highest incidence in East Asia [1]. GC has a low 5-year survival rate in China, with more than 80% of patients being diagnosed at an advanced stage [2]. Traditional treatments for GC patients, such as surgical intervention, cytotoxic therapy, tyrosine kinase inhibitors (TKI), targeting of DNA damage repair proteins, and immunotherapy, have had less than satisfactory results, while new predictive biomarkers and treatment options are urgently needed. Targeted therapy is a therapeutic approach that has emerged in the last decade in the field of anticancer treatment, and it can be applied alone or in combination with conventional chemotherapeutic agents [3, 4]. However, the actual benefits remain relatively low.
The heterogeneity of GC is receiving increasing attention in the course of treatment. Molecular heterogeneity has been shown through the existence of subtypes that have been described on the basis of anatomic site, histopathology and anatomic site, gene expression, gene amplification, DNA methylation, numerous cancer-relevant aberrations and oncogenic pathways [5]. The subtypes were associated with prognosis or clinical outcomes in a limited number of cases.
A growing body of evidence from clinical and laboratory studies suggest that abnormal metabolism is a major hallmark of cancer [6], among which LDH enzyme is one of the important enzymes that shows the disorder of metabolism in different types of cancer [7]. Metabolites are intracellularly active with many genes and proteins, and functional changes in upstream macromolecules (nucleic acids, proteins, etc.) are ultimately reflected at the metabolic level, such as changes in neurotransmitters, hormonal regulation, receptor action effects, cellular signaling release, energy transfer and intercellular communication [8]. Metabolic disorders play a key role in the development and treatment of GC and its prognosis [9, 10]. Studies have shown that proteins associated with lipid metabolism are highly expressed in many cancers [11]. Epidemiological studies have found a higher prevalence of GC in obese populations, while lipid accumulation has been observed in pathological tissues of GC [12]. Chi et al. found that PHTF2-regulated fatty acid metabolism can significantly affect the tumorigenic capacity of GC cells. In addition, glucose metabolism was abnormally elevated in GC patients [13].
Recently, the Cancer Genome Atlas (TCGA) network proposed four GC subtype classification schemes based on the molecular biology of the underlying tumors in each subtype, and the classification study was a milestone. Methods to classify tumors based on genomic atlas such as gene expression, mesenchymal tumor microenvironment and DNA methylation have been continuously explored [14,15,16]. The aim of this study was to investigate the possible clinical significance of metabolism-related features on the prognostic stratification of patients. Two metabolism-related clusters were established through analyzing the gene sets associated with metabolism-related pathways. In addition, we found 6 metabolism-related genes with better performance to predict the subtypes of GC.
Materials and methods
Public sample collection and processing
We systematically collected the gene expression datasets of GC that were publicly available and reported full clinical annotations. Patients without survival information were removed from further evaluation. In total, we gathered six chemo-naïve state cohorts of samples from patients with GC for this study: GSE15459, GSE29272, GSE34942, GSE57303, ACRG/GSE62254, and TCGA-STAD. The raw data from the microarray datasets generated by Affymetrix and Illumina were downloaded from the Gene Expression Omnibus (GEO: https://www.ncbi.nlm.nih.gov/geo/). The raw data from Affymetrix were processed using the RMA algorithm for background adjustment in the Affy software package. RMA was used to perform background adjustment, quantile normalization, and final summarization of oligonucleotides per transcript using the median polish algorithm. The raw data from Illumina were processed using the lumi software package.
Consensus clustering
Consensus clustering was applied to identify potential subtypes of GC according to the expression profiles of metabolism-related pathways by the k-means method. The number of clusters and their stability were determined by the consensus clustering algorithm using the “Consensus Cluster Plus” package. The maximum clustering number was set to six, and the optimal number was determined according to the consensus index and cumulative distribution function (CDF). We performed 100 repetitions to guarantee the stability of our classification.
Immune status analysis among different clusters
We used the CIBERSORT algorithm to quantify the proportions of immune cells in GC [17]. CIBERSORT is a deconvolution algorithm that uses a set of reference gene expression values (a signature with 547 genes) considered a minimal representation for each cell type and, based on those values, infers cell type proportions in data from bulk tumor samples with mixed cell types using support vector regression. We uploaded the normalized gene expression profiles prepared using standard annotation files to the CIBERSORT web portal (https://cibersort.stanford.edu/index.php) using the LM22 signature and 1000 permutations.
Function analysis
Differential analysis was performed using the R package limma to obtain differential genes between the two groups, followed by Gene Ontology (GO) analysis with differentially expressed genes, enrichment of related pathways using the “Cluster Profiler” R package, and visualization in the gene set enrichment analysis (GSEA) of Metascape5. We obtained the GSEA software (version 3.0) from the GSEA website, divided the samples into two groups based on pathway clustering analysis, and downloaded the c2.cp.v7.2.symbols.gmt subset from the Molecular Signatures Database to evaluate the relevant pathways and molecular mechanisms.
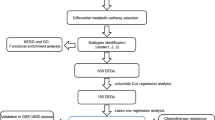
The establishment of a PS-score scoring model
We established an efficient prediction model using the Least absolute shrinkage and selection operator (LASSO) regression model. Overall survival (OS) was then used to derive the most useful predictive features from the training cohort. The “glmnet” package was used to carry out statistical calculations and analysis of the binary logistic regression model. The whole process of data analysis is depicted in Fig. 1. We further implemented stochastic gradient descent in weka3.8.4 software for learning various linear models [binary class Support vector machines (SVM)] and BestFirst (Searches the space of attribute subsets by greedy hill climbing augmented with a backtracking facility) for further filtering of markers. PS-Score = 1.0751*expDLX1 + 3.9879*expFGFR1 + 0.4858*expLTBP2 + 2.7009*expMMP2 − 1.0202*expSLC13A2 − 0.322*expSLCO1B3.

Flow chart of the data analyzing process
Construction of nomogram
The optimal truncation value of the risk score was calculated. For the nomogram, the data of survival time, survival status and 4 features were integrated using the R package rms, and the nomogram was created using the COX method.
ROC analysis of the predict model
The PreScore of each sample was calculated according to the expression level of the sample, and the PreScore distribution of the sample was also characterized. Furthermore, the R package time Receiver operating characteristic (ROC) was used to perform ROC analysis of the PreScore subtype classification.
Quantitative PCR analyses
Total RNA was extracted from patient tumor tissue and adjacent normal tissue using a FastPure® Cell/Tissue Total RNA Isolation Kit V2 (Vazyme). The RNA was reverse transcribed to cDNA using HiScript®IIIRT SuperMix for qPCR (+gDNA wiper) (Vazyme, China), and cDNA products were amplified using a 96-well PCR Plates (Monad) with Taq Pro Universal SYBR qPCR Master mix (Vazyme). The expression of DXL1, FGFR1, LTBP2, MMP2, SLC13A2, SLCO1B3 mRNA in tissue was normalized to that of β-actin. Results were obtained by relative expression ratio (own CT/internal reference CT) (a lower Ct ratio represents a higher expression). The PCR primers sequences are shown in Table 1.
Human gastric tumor tissues collection and processing
The study was approved by the Ethics Committee of the Affiliated Hospital of Nanjing University of Traditional Chinese Medicine (Jiangsu Provincial Hospital of Traditional Chinese Medicine) (No. 2021NL-187-02), and written consent was obtained from all enrolled patients. Gastric tumor tissue from 60 patients came from the Affiliated Hospital of Nanjing University of Traditional Chinese Medicine (Jiangsu Provincial Hospital of Traditional Chinese Medicine), including 60 pairs of gastric tumor tissue and adjacent non-tumor tissue. Inclusion criteria were as follows: (a) histological identification of GC; (b) absence of combined malignancy; and (c) availability of complete follow-up data. Patients who died within 1 month or had received chemotherapy or radiotherapy at the time of gastrectomy were excluded. From 2018 to 2021, 60 samples of gastric tumor tissue and adjacent non-tumor tissue were randomly collected and analyzed by Q-PCR, and patient information is shown in Table 2.
Follow-up
All patients were systematically followed up by trained physicians who adhered to the institutional follow-up program; options for follow-up included outpatient services, letters, telephone calls, mail or visits. Survival time was defined as the time from the date of surgery to the last follow-up visit or date of death.
Survival analysis
Overall survival was performed by R package survival. The survival time, survival status and different immune groups were used to calculate the probability of survival.
Results
Identification of GC molecular subtypes based on metabolism-related pathways
We performed the enrichment scores of 86 human metabolism-related pathways on GC samples to investigate the association with prognosis by Gene Set Variation Analysis (GSVA). The results showed that 34 metabolism-related pathways were significantly associated with prognosis (p value < 0.05; Supplemental Table1). Clustered analysis was also performed in all the samples (GSE57303, GSE34942, GSE84437, ACRG/GSE62254, GSE15459, GSE29272 and TCGA-STAD) through these 34 significant metabolism-related pathways to identify metabolism-related GC subtypes. To characterize the expression pattern of metabolism-related pathways in different GC subtypes, Consensus Cluster Plus—an R package—was used to conduct consistent clustering for the expression profiles, and the maximum cluster number was set to 6. Based on the enrichment score of metabolism-related pathways, the optimal clustering number of 2 was selected (Fig. 2A–C and Supplemental Fig. 1). The enrichment score of metabolism-related pathways in the two subclasses can be seen from the diagram as parts with differences (Fig. 2D). The heatmap showed that cluster 1 (C1) had a high enrichment score in 11 pathways, including diseases of metabolism and diseases associated with glycosaminoglycan metabolism. Cluster 2 (C2) showed high enrichment in 23 pathways, such as metabolism of nucleotides and metabolism of cofactors. We further explored the relationship between our identified GC subtypes and clinical features. Survival analysis (n = 1097) based on TCGA-STAD dataset and 5 GEO datasets (GSE57303, GSE34942, GSE84437, GSE15459 and GSE29272) showed that these two subtypes had significantly different prognoses: C2 had a better prognosis while C1 had a poorer prognosis (log-rank P value < 0.0001, Fig. 2E).

Identification of GC molecular subtypes based on metabolism-related pathways. A Color-coded heatmap corresponding to the consensus matrix for k = 2 obtained by applying consensus clustering. Color gradients represent consensus values from 0 to 1; white corresponds to 0 and dark blue to 1. B Delta area curves for consensus clustering indicating the relative change in area under the cumulative distribution function (CDF) curve for each category number k compared to k-1. The horizontal axis represents the category number k and the vertical axis represents the relative change in area under CDF curve. C Consensus among clusters for each category number k. D Heatmap visualizing of metabolic differences between the two subgroups. E Survival curve of the patients in the two subgroups
Differential gene expression and functional analysis
To further investigate and verify the accuracy of the above results, we applied subtype classification to gene expression data from the Asian Cancer Research Group (ACRG) cohort. We used the R package t test function to evaluate the significance of each gene in the comparison group and control group, and calculated the significant FDR of each gene using the p.adjust function, and finally obtained the difference information of each gene (p value < 0.05, |log2(fold change)|> 1) to finally obtain 528 differentially expressed genes. The differential expression between the two subgroups based on the metabolism-related typing could also be observed by the heat map which suggested that the clusters based on the metabolism-related pathways could be reproduced in other GC cohorts (Fig. 3A). Gene Ontology (GO) enrichment analysis of the differentially expressed genes generated by the two categories described above showed that most of the differentially expressed genes were enriched in extracellular matrix and skeletal muscle-related biological processes (Fig. 3B, C and Supplemental Fig. 2).

Differential gene expression and functional analysis. A The heat map shows that GSE62254 can be divided into two clusters. B, C Circle plot and network visualizing the biological processes enriched by gene ontology (GO) analysis
To elucidate the relationship between these enrichment pathways and clusters, we performed gene set enrichment analysis (GSEA) used to assess the relative expression differences between the two clusters. The results showed that 11 pathways were more highly expressed in cluster1 compared to cluster2 (Supplemental Table 2), while 10 pathways were more highly expressed in cluster2 compared to cluster1 (Supplemental Table 3). Cluster 1 enriched pathways were associated with metastatic spread of GC and their upregulation in expression often predicts a poor prognosis [18, 19]. Cluster 2 enriched pathways were associated with transmembrane transport of substances and glucose metabolism [20], which upregulation of expression of these pathways is positively correlated with a good prognosis. These results were consistent with the analysis of survival differences between cluster 1 and cluster 2.
Patients in the two molecular subtypes exhibited different immune status
Immunotherapy has become an established pillar of anticancer therapy, achieving improved prognosis for a significant number of cancer patients. Therefore, the present study further explored the expression levels of immune cells in metabolism-related subtypes. We totally found six types of immune cells have significantly difference between different GC clusters. The results showed that the expression of fibroblasts, mast cells resting and monocytes in C1 were higher than that in C2. Meanwhile, the activated NK cells, activated memory CD4+ T cells and regulatory T cells were expressed in C2 at higher levels than C1. (Fig. 4). These results suggested that there was an immune infiltration difference between the two subtypes obtained based on metabolism. Immune dysregulation between subtypes may be associated with poor prognosis in patients with GC.

Two molecular subtypes exhibit different immune states. A–F The expression levels of fibroblasts (A) mast cells (B) monocytes (C) NK cells activated (D) T cells CD4+ memory activated (E) and regulatory T cells (F) in C1 and C2 were analyzed using CIBERSORT. G The heatmap of CIBERSORT analysis of six immune cells
Construction of a GC classification model based on metabolism-related clusters
In addition, we performed differential gene analysis between clusters and enriched for relevant marker genes. We used a lasso regression model based on gene expression in the ACRG cohort (300 samples). Random sampling of leave-one-out cross validation was performed for stability and accuracy of results from 528 differentially expressed genes. Finally, 59 genes were selected to construct a diagnostic model \({\sum }_{\mathrm{i}}{\omega }_{\mathrm{i}}{\chi }_{\mathrm{i}}\), where \({\omega }_{\mathrm{i}}\) and \({\chi }_{\mathrm{i}}\) are the coefficient and expression value of each gene, respectively. The cutoff point was 12.53. \({\sum }_{\mathrm{i}}{\omega }_{\mathrm{i}}{\chi }_{\mathrm{i}}\) > 12.53 will be classified into cluster 2 (longer survival), and \({\sum }_{\mathrm{i}}{\omega }_{\mathrm{i}}{\chi }_{\mathrm{i}}\) < 12.53 will be classified into cluster 1 (shorter survival). String 11.0 software was used to infer the interaction between genes, and we found that MMP2, Thrombospondin 1 (THBS1), CLCX12, Versican (VCAN) and TIMP Metallopeptidase Inhibitor 2 (TIMP2) played important roles (Fig. 5A). To further narrow down the scope of the marker, we applied binary class SVM (Implements stochastic gradient descent for learning various linear models) and BestFirst (Studies the space of attribute subsets by greedy hill climbing augmented with a backtracking facility) in Weka3.8.4 software. The 9 markers were filtered using the greedy hill climbing search method with a backtracking facility (Fig. 5B and Supplemental Table 4). Grouping of combinations based on Weka software with nine genes could well distinguish between the two subtypes, and there was a significant survival prognosis difference between the two subtypes (p < 0.0001) (Fig. 5C), and then validated the analysis of each dataset for ROC validation analysis (Fig. 5D). So far, nine marker genes were obtained by screening for differential genes among subtypes obtained based on metabolism.

Construction of a GC classification model based on metabolism-related clusters. A Gene Interaction Network. B LASSO analysis with minimal lambda. C Prognosis after combination of 9 markers. D ROC validation analysis
Screening and clinical validation of marker genes
To explore the relationship between our identified marker genes and clinical features, we performed survival analysis on the ACRG cohort. Among the 9 genes we screened, we found that six marker genes (6/9, 66.7%) were significantly correlated with survival in GC (p value < 0.05). Notably, a high expression level of four markers (DLX1, FGFR1, LTBP2, MMP2) revealed a significantly poor overall survival (OS) (Fig. 6A–D). Meanwhile, a high expression level of two genes (SLC13A2 and SLCO1B3) was associated with a significantly better OS (Fig. 6E, F). Three of these genes were not significantly associated with survival in GC (Fig. 6G–I). Then, we constructed the Nomogram based on the clinical information of the patients and the established risk model (Fig. 6J). The actual model was shown to be largely consistent with the ideal model in the training set GSE26942, indicating that our model has a high accuracy (Fig. 6K, L). In addition, it also showed better predictive power in other validation cohorts (Figure S3).

Survival analysis of marker genes in the classification model. A DLX1. B FGFR1. C LTBP2. D MMP2. E SLC13A2. F SLCO1B3. G-I Genes with no significant differences in survival analysis: APOC3, CPM, POU2F3. J Nomogram integrating risk score and clinical features. K, L Calibration of the nomogram at 3 and 5 years in the training cohort
To test the accuracy of the relationship between the six marker genes and survival prognosis, we verified the expression of the six genes in the tumor tissues of patients. DLX1, FGFR1, LTBP2 and MMP2 were all highly expressed in patients with advanced GC (stage III) (Fig. 7A–D), while SLC12A2 and SLCO1B3 were highly expressed in patients with early GC (stage I and II) (Fig. 7E, F). The Human Protein Atlas showed that the risk genes DLX1, FGFR2, LTBP2, and MMP2 were highly expressed in tumor tissues, while the protein expression levels of the protective genes SLCO1B3 and SLC13A2 were not significantly detected in normal and cancer tissues (Supplemental Fig. 3). Overall, it showed that there were significant differences in the expression of the six screened biomarkers in tumor tissues of patients with different survival stages, and the expression trend of each gene was consistent with the database analysis.

Further validation of prognosis-related genes. A–F Expression of 6 markers in tumor and adjacent normal tissue of GC patients. ***p < 0.001; ****p < 0.0001. G–L Immunohistochemical pictures of paracancerous and cancerous tissues. G DLX1 (Antibody HPA045884). H FGFR1 (Antibody HPA056402). I LTBP2 (Antibody HPA003415). J MMP2 (Antibody CAB002788). K SLC13A2 (Antibody HPA014963). L SLCO1B3 (Antibody HPA050892)
Discussion
In the present study, we identified two molecular subgroups with significant differences in overall survival by consensus clustering. Functional analysis revealed that most of the differentially expressed genes were enriched in extracellular matrix and skeletal muscle-related biological processes. It was shown that extracellular matrix (ECM) remodeling plays an important role in both adipocyte shape/expansion remodeling and skeletal muscle (SM) metabolism [21]. The extracellular matrix not only serves as a tissue scaffold, but also provides key biochemical and biomechanical cues that guide cell growth, survival, migration and differentiation as well as regulate vascular development and immune function [22].
In addition, there was growing evidence that the tumor mesenchyme was an important determinant of cancer progression, and the literature reports that the extracellular matrix contributes to the development and progression of GC [23]. In the present study, GSEA results showed an enrichment of integrin-related pathways with collagen-related pathways in cluster 1. Recent studies have shown that integrin signaling drives multiple stem cell functions, including tumor initiation, epithelial plasticity, metastatic reactivation, and resistance to oncogenes and immune-targeted therapies [24]. The predictive value of αvβ6 integrin in GC has been highlighted, as their increased expression is frequently associated with lymph node metastasis and reduced patient survival in gastrointestinal cancers [25, 26]. Collagen has been shown to be dysregulated in the advanced stages of GC [27, 28]. Notably, a subset of collagen genes are considered to be powerful independent prognostic markers capable of distinguishing precancerous from malignant lesions [29]. The presence of collagen in the tumor microenvironment has been reported to affect the metastasis of cancer cells [19], where COL10A1 has been shown to be a potential inducer of epithelial mesenchymal transition (EMT). Li et al. reported that COL10A1 may play a key role in GC progression and serve as a potential biomarker and therapeutic target for GC patients [30]. The SLC family proteins, PPAR and P53 in the cluster 2 related pathway are associated with the prognosis of GC [31,32,33,34]. The current study shows that pathways enriched in cluster 1 are associated with poor progression of GC, consistent with the poorer survival prognosis of cluster 1. It suggests that GC can be classified into two subtypes based on metabolism.
In addition, we assessed the abundance of immune-associated cells between the two clusters. We found that immune cells highly expressed in cluster 1 were able to secrete a variety of pro-invasive factors [35] or increase vascular permeability [36] to promote tumor invasion and metastasis. Both immune cells enriched in cluster 2 are associated with a good prognosis, for example, NK cells are an important component of the body’s immune surveillance system [37] while CD4+ T cells can both remove tumor cells independently [38] and play an important effect in cellular immunity.
Finally, we screened metabolism-related genes in GC patients to obtain six genes that were significantly associated with prognosis in GC patients. The six genes used for risk modeling in this study have been shown to be strongly associated with tumor development and progression. DLX1 is associated with metastatic status in prostate cancer and is a recognized non-invasive biomarker for prostate cancer (PCa) diagnosis [39]. DLX1 expression is upregulated in high-grade ovarian cancer and promotes the growth and migration/invasion of ovarian cancer cells [40]. In GC, LTBP2 has been shown to promote migration and invasion of GC cells and to predict poor prognosis in GC patients [41]. FGFR1 is a key driver in promoting GC progression and chemoresistance and has been increasingly recognized as a potential therapeutic target for GC [42]. MMP2 has been shown to have a pro-cancer role in hepatocellular carcinoma [43] and is associated with poor prognosis in several cancers [44, 45]. Studies on GC have shown that downregulation of the invasion-related gene MMP2 suppresses invasion and metastasis of gastric cancer [46, 47]. SLC13A2 encodes a transporter protein that transports intermediates of the tricarboxylic acid cycle and plays an important physiological role in the oxidative metabolism of cells [48]. SLCO1B3 of the organic anion transporter family was detected in breast, colon, pancreatic, gastric and prostate cancer cells, among which high expression in breast cancer inhibits the development of breast cancer and can be used as a clinical indicator of prognosis [49]. These reports were consistent with our findings that DLX1, LTBP2, FGFR1 and MMP2 were risk factors for prognosis of GC patients, while SLC13A2 and SLCO1B3 were protective factors. In addition, the nomogram we constructed combined the clinical patient’s gender, age, stage and risk model constructed based on six marker genes, had a more accurate prognosis and could be used as a prognostic tool for GC patients. Secondly, the clinical patient validation results also confirmed that the six marker genes associated with GC metabolism were significantly associated with GC prognosis. Therefore, our results suggested that GC metabolism-related heterogeneity was correlated with the prognosis of GC patients. The risk model constructed based on GC metabolism-related genes is expected to be a survival prediction model for GC. The six GC metabolism-related marker genes may be potential therapeutic targets for GC, and relevant assays can be developed in the future to guide the diagnosis and prognosis of GC clinics.
Conclusions
In conclusion, we staged GC based on metabolism, obtained two metabolically distinct GC subtypes, and further assessed the immune infiltration differences behind these subtypes, which provides additional insights into the relationship between tumor metabolism and immunity. In addition, a survival-related prediction model for GC was constructed based on six screened differential metabolism-related genes in gastric cancer and validated in the ACRG cohort and tissue samples from clinical patients. Our results provide a good theoretical guide for further study of metabolism-related heterogeneity in GC and a target for therapeutic strategies for metabolism-related heterogeneity in GC patients.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. https://doi.org/10.3322/caac.21660.
Zong L, Abe M, Seto Y, Ji J. The challenge of screening for early gastric cancer in China. Lancet. 2016;388(10060):2606. https://doi.org/10.1016/S0140-6736(16)32226-7.
Chen YC, Fang WL, Wang RF, Liu CA, Yang MH, Lo SS, et al. Clinicopathological variation of Lauren classification in gastric cancer. Pathol Oncol Res. 2016;22(1):197–202. https://doi.org/10.1007/s12253-015-9996-6.
Hu B, El HN, Sittler S, Lammert N, Barnes R, Meloni-Ehrig A. Gastric cancer: classification, histology and application of molecular pathology. J Gastrointest Oncol. 2012;3(3):251–61. https://doi.org/10.3978/j.issn.2078-6891.2012.021.
Chia NY, Tan P. Molecular classification of gastric cancer. Ann Oncol. 2016;27(5):763–9. https://doi.org/10.1093/annonc/mdw040.
Park JH, Pyun WY, Park HW. Cancer metabolism: phenotype, signaling and therapeutic targets. Cells Basel. 2020. https://doi.org/10.3390/cells9102308.
Jurisic V, Radenkovic S, Konjevic G. The actual role of LDH as tumor marker, biochemical and clinical aspects. Adv Exp Med Biol. 2015;867:115–24. https://doi.org/10.1007/978-94-017-7215-0_8.
Yang GX, Li X, Snyder M. Investigating metabolite-protein interactions: an overview of available techniques. Methods. 2012;57(4):459–66. https://doi.org/10.1016/j.ymeth.2012.06.013.
Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014;94(2):329–54. https://doi.org/10.1152/physrev.00040.2012.
Xu LB, Zhang HH, Shi MM, Huang ZX, Zhang WT, Chen XD, et al. Metabolic syndrome-related sarcopenia is associated with worse prognosis in patients with gastric cancer: a prospective study. Eur J Surg Oncol. 2020;46(12):2262–9. https://doi.org/10.1016/j.ejso.2020.07.032.
Garber K. Energy deregulation: licensing tumors to grow. Science. 2006;312(5777):1158–9. https://doi.org/10.1126/science.312.5777.1158.
Bourlieu C, Paboeuf G, Chever S, Pezennec S, Cavalier JF, Guyomarc’H F, et al. Adsorption of gastric lipase onto multicomponent model lipid monolayers with phase separation. Colloids Surf B Biointerfaces. 2016;143:97–106. https://doi.org/10.1016/j.colsurfb.2016.03.032.
Yuan LW, Yamashita H, Seto Y. Glucose metabolism in gastric cancer: The cutting-edge. World J Gastroenterol. 2016;22(6):2046–59. https://doi.org/10.3748/wjg.v22.i6.2046.
Ren Q, Zhu P, Zhang H, Ye T, Liu D, Gong Z, et al. Identification and validation of stromal-tumor microenvironment-based subtypes tightly associated with PD-1/PD-L1 immunotherapy and outcomes in patients with gastric cancer. Cancer Cell Int. 2020;20:92. https://doi.org/10.1186/s12935-020-01173-3.
Tahara T, Arisawa T. DNA methylation as a molecular biomarker in gastric cancer. Epigenomics Uk. 2015;7(3):475–86. https://doi.org/10.2217/epi.15.4.
Obradovic J, Todosijevic J, Jurisic V. Application of the conventional and novel methods in testing EGFR variants for NSCLC patients in the last 10 years through different regions: a systematic review. Mol Biol Rep. 2021;48(4):3593–604. https://doi.org/10.1007/s11033-021-06379-w.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7. https://doi.org/10.1038/nmeth.3337.
Xu YH, Li ZL, Qiu SF. IFN-gamma induces gastric cancer cell proliferation and metastasis through upregulation of integrin beta3-mediated NF-kappaB signaling. Transl Oncol. 2018;11(1):182–92. https://doi.org/10.1016/j.tranon.2017.11.008.
Chen D, Liu Z, Liu W, Fu M, Jiang W, Xu S, et al. Predicting postoperative peritoneal metastasis in gastric cancer with serosal invasion using a collagen nomogram. Nat Commun. 2021;12(1):179. https://doi.org/10.1038/s41467-020-20429-0.
Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152. https://doi.org/10.1186/1476-4598-12-152.
Ghanemi A, Melouane A, Yoshioka M, St-Amand J. Secreted protein acidic and rich in cysteine and bioenergetics: extracellular matrix, adipocytes remodeling and skeletal muscle metabolism. Int J Biochem Cell Biol. 2019;117: 105627. https://doi.org/10.1016/j.biocel.2019.105627.
Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. Embo Rep. 2014;15(12):1243–53. https://doi.org/10.15252/embr.201439246.
Moreira AM, Pereira J, Melo S, Fernandes MS, Carneiro P, Seruca R, et al. The extracellular matrix: an accomplice in gastric cancer development and progression. Cells Basel. 2020. https://doi.org/10.3390/cells9020394.
Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35(3):347–67. https://doi.org/10.1016/j.ccell.2019.01.007.
Kawashima A, Tsugawa S, Boku A, Kobayashi M, Minamoto T, Nakanishi I, et al. Expression of alphav integrin family in gastric carcinomas: increased alphavbeta6 is associated with lymph node metastasis. Pathol Res Pract. 2003;199(2):57–64. https://doi.org/10.1078/0344-0338-00355.
Lian PL, Liu Z, Yang GY, Zhao R, Zhang ZY, Chen YG, et al. Integrin alphavbeta6 and matrix metalloproteinase 9 correlate with survival in gastric cancer. World J Gastroenterol. 2016;22(14):3852–9. https://doi.org/10.3748/wjg.v22.i14.3852.
Jiang X, Wu M, Xu X, Zhang L, Huang Y, Xu Z, et al. COL12A1, a novel potential prognostic factor and therapeutic target in gastric cancer. Mol Med Rep. 2019;20(4):3103–12. https://doi.org/10.3892/mmr.2019.10548.
Zhang QN, Zhu HL, Xia MT, Liao J, Huang XT, Xiao JW, et al. A panel of collagen genes are associated with prognosis of patients with gastric cancer and regulated by microRNA-29c-3p: an integrated bioinformatics analysis and experimental validation. Cancer Manag Res. 2019;11:4757–72. https://doi.org/10.2147/CMAR.S198331.
Zhao Y, Zhou T, Li A, Yao H, He F, Wang L, et al. A potential role of collagens expression in distinguishing between premalignant and malignant lesions in stomach. Anat Rec (Hoboken). 2009;292(5):692–700. https://doi.org/10.1002/ar.20874.
Li T, Huang H, Shi G, Zhao L, Li T, Zhang Z, et al. TGF-beta1-SOX9 axis-inducible COL10A1 promotes invasion and metastasis in gastric cancer via epithelial-to-mesenchymal transition. Cell Death Dis. 2018;9(9):849. https://doi.org/10.1038/s41419-018-0877-2.
Ding B, Lou W, Xu L, Li R, Fan W. Analysis the prognostic values of solute carrier (SLC) family 39 genes in gastric cancer. Am J Transl Res. 2019;11(1):486–98.
Sato H, Ishihara S, Kawashima K, Moriyama N, Suetsugu H, Kazumori H, et al. Expression of peroxisome proliferator-activated receptor (PPAR)gamma in gastric cancer and inhibitory effects of PPARgamma agonists. Br J Cancer. 2000;83(10):1394–400. https://doi.org/10.1054/bjoc.2000.1457.
Wagner N, Wagner KD. PPAR beta/delta and the hallmarks of cancer. Cells Basel. 2020. https://doi.org/10.3390/cells9051133.
Kim KW, Kim N, Choi Y, Kim WS, Yoon H, Shin CM, et al. Different effects of p53 protein overexpression on the survival of gastric cancer patients according to Lauren histologic classification: a retrospective study. Gastric Cancer. 2021;24(4):844–57. https://doi.org/10.1007/s10120-021-01163-y.
Han Y, Zhang Y, Jia T, Sun Y. Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts. Tumour Biol. 2015;36(3):1385–94. https://doi.org/10.1007/s13277-015-3230-8.
Noto CN, Hoft SG, DiPaolo RJ. Mast cells as important regulators in autoimmunity and cancer development. Front Cell Dev Biol. 2021;9: 752350. https://doi.org/10.3389/fcell.2021.752350.
Du Y, Wei Y. Therapeutic potential of natural killer cells in gastric cancer. Front Immunol. 2018;9:3095. https://doi.org/10.3389/fimmu.2018.03095.
Wei M, Shen D, Mulmi SS, Liu J, Zhang J, Yin Y. The progress of T cell immunity related to prognosis in gastric cancer. Biomed Res Int. 2018;2018:3201940. https://doi.org/10.1155/2018/3201940.
Goel S, Bhatia V, Kundu S, Biswas T, Carskadon S, Gupta N, et al. Transcriptional network involving ERG and AR orchestrates Distal-less homeobox-1 mediated prostate cancer progression. Nat Commun. 2021;12(1):5325. https://doi.org/10.1038/s41467-021-25623-2.
Chan DW, Hui WW, Wang JJ, Yung MM, Hui LM, Qin Y, et al. DLX1 acts as a crucial target of FOXM1 to promote ovarian cancer aggressiveness by enhancing TGF-beta/SMAD4 signaling. Oncogene. 2017;36(10):1404–16. https://doi.org/10.1038/onc.2016.307.
Wang J, Liang WJ, Min GT, Wang HP, Chen W, Yao N. LTBP2 promotes the migration and invasion of gastric cancer cells and predicts poor outcome of patients with gastric cancer. Int J Oncol. 2018;52(6):1886–98. https://doi.org/10.3892/ijo.2018.4356.
Chen Q, Zhu M, Xie J, Dong Z, Khushafah F, Yun D, et al. Design and synthesis of novel nordihydroguaiaretic acid (NDGA) analogues as potential FGFR1 kinase inhibitors with anti-gastric activity and chemosensitizing effect. Front Pharmacol. 2020;11: 518068. https://doi.org/10.3389/fphar.2020.518068.
Liu D, Kang H, Gao M, Jin L, Zhang F, Chen D, et al. Exosome-transmitted circ_MMP2 promotes hepatocellular carcinoma metastasis by upregulating MMP2. Mol Oncol. 2020;14(6):1365–80. https://doi.org/10.1002/1878-0261.12637.
Gobin E, Bagwell K, Wagner J, Mysona D, Sandirasegarane S, Smith N, et al. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer. 2019;19(1):581. https://doi.org/10.1186/s12885-019-5768-0.
Han L, Sheng B, Zeng Q, Yao W, Jiang Q. Correlation between MMP2 expression in lung cancer tissues and clinical parameters: a retrospective clinical analysis. Bmc Pulm Med. 2020;20(1):283. https://doi.org/10.1186/s12890-020-01317-1.
Wang J, Cai H, Xia Y, Wang S, Xing L, Chen C, et al. Bufalin inhibits gastric cancer invasion and metastasis by down-regulating Wnt/ASCL2 expression. Oncotarget. 2018;9(34):23320–33. https://doi.org/10.18632/oncotarget.24157.
Sun L, Zhang Y, Lou J. ARHGAP9 siRNA inhibits gastric cancer cell proliferation and EMT via inactivating Akt, p38 signaling and inhibiting MMP2 and MMP9. Int J Clin Exp Pathol. 2017;10(12):11979–85.
Bergeron MJ, Clemencon B, Hediger MA, Markovich D. SLC13 family of Na(+)-coupled di- and tri-carboxylate/sulfate transporters. Mol Aspects Med. 2013;34(2–3):299–312. https://doi.org/10.1016/j.mam.2012.12.001.
Tang T, Wang G, Liu S, Zhang Z, Liu C, Li F, et al. Highly expressed SLCO1B3 inhibits the occurrence and development of breast cancer and can be used as a clinical indicator of prognosis. Sci Rep. 2021;11(1):631. https://doi.org/10.1038/s41598-020-80152-0.
Acknowledgements
Not applicable.
Funding
This study was supported by National Natural Science Foundation of China (No.81973782, No.81704031), Science and Technology Planning Project of Jiangsu Province, China (No. BK20211392), Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX22_0749, SJCX21_0692, SJCX21-0740), The Nanjing Medical Science and Technique Development Foundation (ZKX19022), Jiangsu Provincial High level health talent “six one project” (LGY2019005).
Author information
Authors and Affiliations
Contributions
XXZ, XC, YQL, JW, MLC, RJZ, XTX, QMS and TYX: jointly designed the study and drafted the manuscript. JYL: was responsible for the collection of clinical specimens. JW, QMS, and TYX: supervised the study. All the authors contributed to the data collection, analysis and interpretation, manuscript writing and revision. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Ethics approval
This retrospective observational study involving human participants was in accordance with the ethical standards of the institutional research committee. The Ethics Committee of the Affiliated Hospital of Nanjing University of Traditional Chinese Medicine (Jiangsu Provincial Hospital of Traditional Chinese Medicine) approved this study (No. 20221NL18702).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, X., Chen, X., Liu, J. et al. A novel metabolism-related prognostic gene development and validation in gastric cancer. Clin Transl Oncol 25, 447–459 (2023). https://doi.org/10.1007/s12094-022-02958-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-022-02958-w