
Abstract
Actions of cismethrin versus deltamethrin were compared using two functional attributes of rat brain synaptosomes. Both pyrethroids increased calcium influx but only deltamethrin increased Ca2+-dependent neurotransmitter release following K+-stimulated depolarization. The action of deltamethrin was stereospecific, concentration-dependent, and blocked by ω-conotoxin GVIA. These findings delineate a separate action for deltamethrin and implicate N-type rat brain Cav2.2 voltage-sensitive calcium channels (VSCC) as target sites that are consistent with the in vivo release of neurotransmitter caused by deltamethrin. Deltamethrin (10−7 M) reduced the peak current (approx. −47%) of heterologously expressed wild type Cav2.2 in a stereospecific manner. Mutation of threonine 422 to glutamic acid (T422E) in the α1-subunit results in a channel that functions as if it were permanently phosphorylated. Deltamethrin now increased peak current (approx. +49%) of T422E Cav2.2 in a stereospecific manner. Collectively, these results substantiate that Cav2.2 is directly modified by deltamethrin but the resulting perturbation is dependent upon the phosphorylation state of Cav2.2. Our findings may provide a partial explanation for the different toxic syndromes produced by these structurally-distinct pyrethroids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Role of voltage-sensitive calcium channels in pyrethroid neurotoxicity
It is well established that voltage-sensitive sodium channel isoforms are modified by pyrethroids (Narahashi 1992; Trainer et al. 1997). There is emerging evidence, however, suggesting that other target sites may also be involved with the acute neurotoxic effects of pyrethroids (Soderlund et al. 2002). Several extensive reviews of the toxicological ramifications at these receptor sites exist (Clark 1994; Narahashi 1992; Soderlund and Bloomquist 1989; Soderlund et al. 2002). Of the potential target sites implicated in the action of pyrethroids, only voltage-sensitive sodium, calcium and chloride channels are altered by relatively low concentrations of pyrethroids, elicit stereospecific actions, and have been implicated in the acute neurotoxicological response using functional assays (Soderlund et al. 2002).
Two main classes of pyrethroids have been characterized based on their observed acute neurotoxicological symptoms in mammals. In general, pyrethroids that induce a tremor response are T-syndrome pyrethroids and pyrethroids that induce a choreoathetosis with salivation response are CS-syndrome pyrethroids. However, not all pyrethroids fall neatly into either of these two categories (Soderlund et al. 2002).
Early acute toxicity studies indicated that the in vivo action of CS-syndrome pyrethroids on the nervous system was different than that of T-syndrome pyrethroids. Deltamethrin (CS-syndrome pyrethroid) caused a 52% decrease in acetylcholine content of the cerebellum, whereas DDT, a well-established voltage-sensitive sodium channel agonist, and cismethrin, a T-syndrome pyrethroid, caused no significant reduction (Aldridge et al. 1978). An effect of deltamethrin on neurotransmitter release has recently been validated in freely moving rats exhibiting the CS symptoms (Hossain et al. 2004) where acetylcholine release from the hippocampus was enhanced dose dependently up to more than fivefold in rats exposed to deltamethrin at the onset of the CS-symptoms. These in vivo experiments delineate a physiological response (enhanced neurotransmitter release) that is different between cismethrin versus deltamethrin, which produce the T- and CS-syndromes, respectively, and suggest that other mechanisms, beside voltage-sensitive sodium channel agonism, may be involved with the neurotoxic action of CS-syndrome pyrethroids.
Release of neurotransmitter from presynaptic nerve terminals following action potential depolarization occurs in a rapid and highly regulated manner (Catterall 1998). Neurotransmitter release is triggered by voltage-sensitive Ca2+ entry, via N- and P/Q-type VSCC, during depolarization (Turner et al. 1993). Neurotransmitters are released in a quantal fashion (Katz and Miledi 1967) by fusion of synaptic vesicles with the synaptolemma at specific loci (active release zones) (Koenig et al. 1998). The biophysical properties and organization of the presynaptic nerve terminal is critical for rapid and highly-localized delivery of external Ca2+ to the intracellular neurotransmitter releasing machinery (Catterall 1999). The resulting brief rise in the intracellular Ca2+ concentration to the relatively high level necessary for exocytosis (∼1–3 mM) occurs only in close proximity to specific VSCC that are co-assembled in active release zones (Harlow et al. 2001). This arrangement is necessary to minimize the cytotoxicity of high levels of intacellular free Ca2+.
Early in vitro findings established that deltamethrin stimulates the spontaneous release of [3H]GABA from mammalian synaptosomes superfused with non-depolarizing saline buffer and this release was substantially tetrodotoxin (TTX)-sensitive (Eells and Dubocovich 1988; Nicholson et al. 1987). In a comprehensive analysis of 25 pyrethroids, Doherty et al. (1987) found that most of the pyrethroids examined increased the Na+-dependent release of neurotransmitters from rat brain synaptosomes but the release was only partially abolished by TTX.
Neurotransmitter release induced by elevating external K+ concentrations is a more physiologically relevant means to investigate pyrethroid effects on action potential-induced, Ca2+-dependent, neurotransmitter release since this process has been shown to open Cav2.2, allowing Ca2+ influx and subsequent Ca2+-triggered neurotransmitter release (Meder et al. 1999). Furthermore, K+-stimulated Ca2+ uptake and subsequent neurotransmitter release in synaptosomes is blocked by a variety of VSCC antagonists, but not by TTX (Turner et al. 1993; Meder et al. 1999; Fink et al. 2002).
CS-syndrome pyrethroids increased Ca2+-dependent norepinephrine release from rat brain synaptosomes depolarized by high K+ isolated while T-syndrome pyrethroids were much less potent and efficacious in evoking release (Brooks and Clark 1987). Deltamethrin, a CS-syndrome pyrethroid, still increased release in the presence of TTX but release was inhibited by the specific calcium channel blocker, D595. Deltamethrin was also a potent agonist of Ca2+-dependent neurotransmitter release from synaptosomes of a variety of organisms (Clark and Brooks 1989b; Clark and Marion 1990; Guo-lei et al. 1992).
Deltamethrin was also found to be highly toxic to Paramecium tetraurelia, an organism that does not possess a voltage-sensitive sodium channel. Toxicity was concentration-dependent, stereospecific and enhanced under K+-stimulated depolarizing conditions (Clark et al. 1995). Paramecium backward swimming behavior, a cellular response controlled by ciliary VSCC (Ehrlich et al. 1988), was increased by deltamethrin in a concentration-dependent and stereospecific manner and was highly correlated to deltamethrin toxicity. Pawn mutants, which are incapable of backward swimming due to non-functioning ciliary VSCC, were completely unaffected by deltamethrin (Clark et al. 1995). Using a variety of fluorescent and radioisotope techniques under depolarizing conditions, deltamethrin resulted in increased Ca2+ influx that was concentration-dependent and blocked by D595 (Symington et al. 1999a). Thus, CS-syndrome pyrethroids, specifically deltamethrin, are potent agonists of the ciliary VSCC in P. tetraurelia and induce death by osmotic lysis in the absence of a voltage-sensitive sodium channel (Symington et al. 1999b).
A similar action of deltamethrin has been reported in housefly thoracic ganglia. A low voltage-activated VSCC was modified by deltamethrin resulting in a hyperpolarizing shift of the activation midpoint potential (Duce et al. 1999a). In fluorescence experiments using the same preparation, deltamethrin stimulated Ca2+ influx, which was inhibited by VSCC blockers but was unaltered by TTX (Duce et al. 1999b).
Contrary to the above, pyrethroids have been shown to block distinct classes of VSCC in a variety of non-neuronal mammalian systems. In mouse neuroblastoma cells (N1E-115), tetramethrin, a T-syndrome pyrethroid, preferentially blocked T-type calcium current by 75% but only 30% of the L-type current (Narahashi et al. 1987; Yoshii et al. 1985). Tetramethrin also blocked a T-type calcium channel current in rabbit sino-atrial node cells. Recently, electrophysiological studies using Cav3.1 (T-type), Cav1.2 (L-type), and Cav2.1 (P/Q-type) expressed in non-neuronal HEK cells indicated that bioallethrin, a T-syndrome pyrethroid, blocked all three channels (Hildebrand et al. 2004).
Collectively, these findings substantiate that VSCC are modified by pyrethroids. The mechanisms by which T- and CS-syndrome pyrethroids accomplish this, however, may be different. Specifically, these results indicate that additional sites of action for deltamethrin, and perhaps other CS-syndrome pyrethroids, may include Cav2 and Cav3 channels associated with the synaptolemma of presynaptic nerve terminals from the CNS.
Rat brain Cav2.2 as a site of action for deltamethrin
Recent electrophysiological studies with rat brain Cav2.2 expressed in Xenopus laevis oocytes substantiated that deltamethrin modified Cav2.2 in vitro (Symington and Clark 2005). Deltamethrin reduced Ba2+ peak current in a concentration-dependent and stereospecific manner, caused a hyperpolarizing shift in the midpoint potential of activation and slowed both activation and inactivation rates. Eventhough the amplitude of the peak current is reduced, the total ion current may actually be increased due to the combined effects of the hyperpolarizing shift in the activation curve and the slowing of inactivation. Regardless, these in vitro experiments directly validate that the operation of Cav2.2, the VSCC most responsible for neurotransmitter release, are altered by deltamethrin.
The inhibition of Ba2+ peak current by deltamethrin via heterologously expressed Cav2.2 may nonetheless be inconsistent with increased Ca2+ influx and enhanced neurotransmitter release reported using a functional synaptosomal preparation (Symington and Clark 2005) and with the in vivo observations described previously (Aldridge et al. 1978; Hossain et al. 2004). Such differences suggest that other regulatory components modulating Ca2+ influx and subsequent neurotransmitter release, such as calmodulin binding or channel phosphorylation (Catterall 1997, 1998), may be necessary to elicit a neurophysiologic response that is consistent with the in vivo symptoms of deltamethrin poisoning but are likely absent in non-neuronal heterologous expression systems such as Xenopus oocytes. If this is the case, electrophysiological measurements preformed using Ba2+ as a charge carrier would not activate Ca2+-dependent processes (calmodulin binding, Ca2+-dependent phosphorylation, etc.) and may not be the most realistic means to determine the action of neurotoxic pyrethroids on VSCC.
VSCC are directly modulated by phosphorylation/dephosphorylation events involving protein kinases and phosphatases, respectively (Rossie 1999). The specific effect of phosphorylation is highly dependent on the channel/tissue type that is being analyzed. Using rat brain synaptosomes, Nichols et al. (1987) reported that phorbol esters stimulated protein kinase C (PKC), which increased Ca2+-dependent neurotransmitter release under K+-stimulated depolarization conditions. More recently, the direct modulatory effect of PKC on Cav2.2 has been demonstrated (Stea et al. 1995). Collectively, these results suggest that PKC-dependent phosphorylation of Cav2.2 increases Ca2+ influx and subsequent enhanced neurotransmitter release.
The α1-subunit of Cav2.2 (α1B) possesses serine/threonine residues that can be phosphorylated by protein kinase A, C, and G (PKA, PKC, PKG, respectively). Addition of the phorbol ester, 12-myristate 13-acetate (PMA), enhanced the current amplitude of Cav2.2 expressed in Xenopus oocytes (Stea et al. 1995), suggesting that Cav2.2 is up regulated by PKC-dependent phosphorylation. Furthermore, over expression of protein phosphatase 2cα in tsA cells expressing Cav2.2 significantly decreased phorbol dibutyrate (PDBu)-induced Ca2+ current amplitude (Li et al. 2005).
An intricate model exists for the precise control of neurotransmitter release at nerve terminals involving a variety of intracellular signal mediators that converge at the α1B-subunit of Cav2.2 (Hamid et al. 1999). The region between the first and second domains of the α1B-subunit (DI-DII region) has overlapping binding sites for PKC, betagamma subunit of heterotrimeric G-proteins (Gβγ) and the VSCC β-subunit. PKC-dependent phosphorylation of the α1B-subunit up regulates Cav2.2 but only in the combined presence of a β-subunit (Stea et al. 1995). PKC-dependent phosphorylation of either threonine 422 (T422) or serine 425 (S425) in the α1B-subunit up regulates Cav2.2 by shifting the channel to a “willing state” (De Waard et al. 1997). Conversely, Gβγ binding to the DI-DII region of the α1B-subunit of Cav2.2 inhibits the channel by shifting it to the “reluctant state” (Zamponi et al. 1997). Phosphorylation of T422 antagonizes Gβγ-dependent inhibition (Zamponi et al. 1997).
PMA up-regulation of Cav2.2 is mimicked by amino acid substitutions in the PKC regulatory site within the intracellular loop between DI-DII of the α1B-subunit. Conversion of threonine 422 to glutamic acid (T422E) results in a channel that functions as if it were permanently phosphorylated since glutamic acid is negatively charged at physiological pH (Hamid et al. 1999; Cooper et al. 2000). This mutation produces a phosphoform of Cav2.2 that mimics PMA up-regulation (it acts as if tonically phosphorylated) and results in a channel that is no longer sensitive to Gβγ inhibition. Thus, T422 acts as a molecular switch to regulate multiple convergent signal transductions systems within the α1-subunit of Cav2.2 (Hamid et al. 1999; Cooper et al. 2000).
Deltamethrin has previously been shown to modify phosphorylation cascades in the presynaptic nerve terminal of invertebrates (Matsumura et al. 1989; Miyazawa and Matsumura 1990; Osborne et al. 1995) and vertebrates (Ishikawa et al. 1989; Enan and Matsumura 1991, 1993; Kanemoto et al. 1992). Specifically, deltamethrin was found to stimulate the PKC/phosphoinositide pathway in rat brain synaptosomes, which increased intracellular 1,4,5-triphosphate (IP3) and free Ca2+ (Enan and Matsumura 1993). Together, these results implicate a role for phosphorylation in the neurotoxic action of deltamethrin, which may be responsible for the reported differences noted between our functional synaptosomal assays and those obtained in vivo versus those obtained using electrophysiological responses from heterologously expressed VSCC.
In this paper, the actions of a classic T-syndrome (cismethrin) and CS-syndrome (deltamethrin) pyrethroid are evaluated using two functional attributes of rat brain synaptosomes; Ca2+ influx and endogenous neurotransmitter release (L-glutamate) following K+-stimulated depolarization. It is hypothesized that pyrethroids that exert different syndromes will also exert different effects on these functional attributes. If true, alteration of such important physiological processes at presynaptic nerve terminals would likely impact the acute neurotoxicological response caused by these pyrethroids. Additionally, electrophysiological studies are carried out using rat brain Cav2.2 expressed in Xenopus oocytes to establish a direct action of deltamethrin on a N-type VSCC, the channels most responsible for Ca2+-dependent neurotransmitter release during action potential depolarization. Lastly, site-directed mutagenesis is used to replace threonine 422 with glutamic acid (T442E), which produces a mutant that functions as a permanently phosphorylated channel. It is hypothesized that alteration of this PKC-dependent phosphorylation site will modify the action of deltamethrin on Cav2.2 in a manner that this consistent with enhanced neurotransmitter release, a hallmark of the action of deltamethrin in vivo.
Materials and methods
Materials
Technical grade pyrethroids, 1R-deltamethrin ((S)-α-cyano-3-phenoxybenzyl (1R,3R)-3-(2,2-dibromovinyl)-2,2-dimethylcyclopropanecarboxylate) (98% pure, lot # 2N0746B(R92-2040)) and bioresmethrin (5-benzyl-3-furylmethyl (1R,3R)-2,2-dimethyl-3-(2-methylprop-1-enyl)cyclopropanecarboxylate) (93.3% pure, lot # 8N304B(R99-0950)), were provided by the Pyrethroid Working Group (PWG)1. Cismethrin (5-benzyl-3-furylmethyl (1R,3S)-2,2-dimethyl-3-(2-methylprop-1-enyl)cyclopropanecarboxylate) was purified (99.8%) according to previously reported methods (Bloomquist and Soderlund 1988). The inactive 1S-enantiomer of deltamethrin (>99% pure) was provided by Dr. D.M. Soderlund (Cornell University). Pyrethroid stock solutions (10−14–10−2 M) were prepared in dimethylsulfoxide (DMSO, 0.2% final assay concentration) as a solvent and diluted as required.
Ex-breeder female Sprague-Dawley rats (400–600 g) were purchased from Charles River Laboratories (Boston, MA, USA) and all animal procedures were conducted in accordance with IACUC guidelines (Protocol No. 23-09-09R).
Percoll was purchased from Amersham Biosciences (Piscataway, NJ). Fura-2 (oxazolecarboxylic acid, 2-(6-(bis(2-((acetyloxy)methoxy)-2-oxoethyl)amino)-5-(2-(2-(bis(2-((acetyloxy)methoxy)-2-oxoethyl)amino)-5-methylphenoxy)ethoxy)-2-benzofuranyl))-acetylmethoxyl ester and Amplex Red reagent (10-acetyl-3,7-dihydroxyphenoxazine) were purchased from Molecular Probes (Eugene, OR, USA). Tetrodotoxin (TTX) and ω-conotoxin GVIA (GVIA) were purchased from Alomone Laboratory (Jerusalem, Israel). All other chemicals were purchased from the Sigma Chemical Co. (St. Louis, MO, USA) at the highest purity available.
Synaptosome preparation
Synaptosomes were prepared by homogenization of whole rat brain sans brain stem and purified on a discontinuous Percoll gradient as previously described (Symington and Clark 2005). Protein concentrations were determined using the bicinchoninic acid method (Smith et al. 1985) with bovine serum albumin (BSA) as a standard protein.
Ca2+ influx assay
A fura-2 AM fluorescent assay was used to measure Ca2+ influx as previously described (Zhang 1996; Zhang and Nicholson 1994; Symington and Clark 2005). Fura-2 loaded synaptosomes, pretreated with pyrethroids and ion channel toxins (see section below), were diluted into saline B buffer, aliquoted to a 96-well plate and fluorescence monitored (Exλ = 340 nm and 380 nm and Emλ = 510 nm) at 37°C using a Gemini-XS fluorescent microplate reader (Molecular Devices, Carlsbad, CA, USA) equipped with Softmax Pro program (ver. 3.1.2) to determine the basal fluorescence. Synaptosomes were subsequently depolarized by the addition of a 5 μl aliquot of KCl (final concentration range = 0–60 mM), incubated for additional 2 min, and fluorescence recorded to determine the increase in the internal free Ca2+ concentration ([Ca2+]i). Values reported are change (Δ) in [Ca2+]i/μg protein due depolarization following treatments as calculated by Eq. 1 (Symington and Clark 2005).
Fura-2 calibrations and [Ca2+]i calculations were performed as described elsewhere (Grynkiewicz et al. 1985; Iredale and Dickenson 1995).
Endogenous neurotransmitter release assay
Endogenous neurotransmitter release was determined using l-glutamate detection via an enzyme-linked assay with Amplex Red Reagent™ as the fluorophore (Nicholls et al. 1987; Zhang 1996; Symington and Clark 2005). Synaptosomes in saline A with glucose were treated as for the Ca2+ influx determinations except that 50 μM Amplex, 0.04 U/ml glutamate oxidase and 0.125 U/ml horse-radish peroxidase were added to saline B and aliquoted to a 96-well assay plate. Basal fluorescence was monitored (Exλ = 530 nm and Emλ = 591 nm) as before. Synaptosomes were depolarized with KCl and fluorescence recorded for an additional 30 min post depolarization to determine the net increase in the amount of l-glutamate released. The 30 min incubation assures quantitative detection of released l-glutamate (Molecular Probes Product Information MP 12221) that occurs over the first ∼10 s of depolarization (Wennemuth et al. 2000). Total l-glutamate content of synaptosomes was determined as above, but in the presence of 0.5% Triton X-100. Percent glutamate release values reported are of total glutamate released (TGR) due to depolarization following treatment as a function of the total synaptosomes glutamate content (released by Triton X-100 treatment) as calculated by Eq. 2.
Pretreatments with pyrethroids and ion channel toxins
Synaptosomes (100 μl) were pretreated with 0.2 μl of a 1000-fold concentrated stock solution of pyrethroids in DMSO or DMSO alone (0.2%, final concentration) and incubated at 37°C for 20 min prior to the start of each assay. For ion channel toxin pretreatments, appropriate concentrations of each ion channel toxin were added 5 min prior to the addition of pyrethroid and incubated at 37°C.
Statistical analysis of biochemical data
The effects of increasing concentrations of pyrethroids were assessed for each of the functional assays as described in Eqs. 1 and 2. Pyrethroid concentrations ranged from 2 × 10−17 to 2 × 10−5 M. Individual responses for each concentration were determined from an average of eight replicates per synaptosome preparation and concentration-dependent response curves were generated from multiple synaptosomes preparations (n ≥ 3).
The concentration-dependent response data from each assay were fitted to the Hill equation using GraphPad Prism software (version 3.00 for Windows, GraphPad Software, San Diego California USA) by minimizing the sum-of-squares. Relative indices of binding (Hill slope), potency (Log EC50), and efficacy (βmax) were calculated from the sigmoidal fit of the data using the Hill equation (Symington and Clark 2005). Statistical significance for each of the indices due to treatment was assessed by ANOVA (GraphPad Prism) with a Newman-Keuls Multiple Comparison Post Hoc Test using P < 0.05 as the criteria for evaluation. An F-test calculated from the sum of the squared residuals was used to compare the sigmoid fits of the concentration-response data for each of the functional assays (MS-Excel).
Site directed mutagenesis
Construction of mutant Cav2.2 α1-subunit possessing the T422E alteration was performed using the Quick Change Site-Directed Mutagenesis Kit according to the manufacture’s instructions (Stratagene, La Jolla, CA, USA). A mutagenic oligonucleotide primer set was designed based on the sequence of the α1-subunit of Cav2.2. The T422E forward primer was 5′GTTGAAGAGAGCTGCTGAGAAGAAGAGCCGAAATGACC and the T422E reverse primer was 5′CATTTCGGCTCTTCTTCTCAGCAGCTCTCTTCAACACA.
The cDNA encoding the ORF of Cav2.2 α1-subunit (α1B-d) was cloned into One Shot Top10F’ E. coli cells (Invitrogen, Carlsbad, CA, USA). New double-stranded DNA plasmids containing the mutation of interest were synthesized by nonstrand-displacing Pfu Turbo DNA polymerase using the mutagenic oligonucleotide primer set and the cDNA of the wild-type (non-mutated) α1-subunit of Cav2.2 as template. The methylated parental plasmid DNA (without mutation) was digested by the endonuclease Dpn I (10 U/μl) at 37°C for 1 h according to the manufacturer’s instructions. Mutant Cav2.2 α1-subunit vector was subcloned into XL10-Gold competent cells. The success of mutagenesis was confirmed by automatic sequencing using ABI Prism 3700 DNA Analyzer (PE Applied Biosystems) by the National Instrumentation Center for Environmental Management at Seoul National University (Seoul, Korea). Mutant Cav2.2 α1-subunit vector was purified and the linearized plasmid used in synthesis of cRNA as described below.
Heterologous expression of Cav2.2 and electrophysiology
Full length clones for the Cav2.2 α1 and β3 subunits were coexpressed as previously described (Lin et al. 1997; Symington and Clark 2005). The cRNA transcripts were synthesized from linearized plasmid containing the α1 and β3 cDNAs using the mMessage mMachine™ in vitro transcription kit according to the manufacturer’s instructions (Ambion, Austin, TX, USA). Oocytes were co-injected with 25–50 nl of α1 (180 ng/μl) and β3 (60 ng/μl) subunit cRNAs (Soreq and Seidman 1992) and incubated at 19°C for 2–5 days prior to electrophysiological recordings.
The functional attributes of expressed Cav2.2 were assayed using the two-electrode voltage clamp (TEVC) technique by measuring inward Ba2+ currents (Lin et al. 1997). Data were digitized at 20 kHz using the Digidata 1322A digitizer, stored by the pClamp (ver. 8.2, Axon Instruments, Union City, CA, USA) software, and capacitive transient current subtracted online using the P4 protocol (Lin et al. 1997). Pulse protocols were initiated from a holding potential of −80 mV to a test potential of 0 mV for 2.5 s to determine currents under steady-state depolarization. Pulse protocols were initiated from a holding potential of −80 mV to a final potential of +40 mV in 5 mV steps, each for 150 ms, to determine currents used for current-voltage relationships. Additional details of the pulse protocols are described in specific figure legends. Analysis of the electrophysiology data used a combination of Clampfit 8.2 (ver 8.2, Axon Instruments) and GraphPad Prism following the procedures previously described (Symington and Clark 2005).
Results
Effects of cismethrin and deltamethrin on calcium influx and glutamate release
Synaptosomes depolarized in the presence of external free Ca2+ elicited a K+-stimulated Ca2+ influx, a hallmark of functional synaptosomes (Fig. 1a). This response did not occur in the absence of external Ca2+ and was eliminated by the addition of the Ca2+ chelator, EGTA, to the buffer.

The effect of cismethrin or deltamethrin on K+-stimulated Ca2+ influx into rat brain synaptosomes. a K+-stimulated Ca2+ influx is dependent on external Ca2+. b The effect of increasing K+ concentration on pyrethroid-dependent Ca2+ influx. An open triangle indicates that [Ca2+]i is significantly lower after treatment with 60 mM K+ (unpaired t test, P < 0.05). A closed diamond indicates that [Ca2+]i is significantly higher after treatment with 60 mM K+ (n = 8, unpaired t test, P < 0.05)
Cismethrin (2 × 10−7 M) exhibited no effect on Ca2+ influx when depolarized with low to moderate K+ concentrations (< 20 mM) (Fig. 1b). At K+ concentrations of 20 mM or greater, cismethrin significantly increased Ca2+ influx by an average of 1.5-fold (±0.08) (average was calculated from the fold increases in Ca2+ influx evoked by cismethrin compared to DMSO at each individual K+ concentration) compared to synaptosomes treated with only DMSO (paired t test, P = 0.0005). Deltamethrin (2 × 10−7 M) elicited significantly more Ca2+ influx compared to synaptosomes treated only with DMSO at all K+ concentrations and resulted in an average increase of 1.8-fold (±0.06) (paired t test, P < 0.0001, Fig. 1b).
Cismethrin (toxic 1R-cis stereoisomer) elicited an average 1.6-fold (±0.20) increase in Ca2+ influx at moderate (20 mM) and high (60 mM) K+ concentrations compared to DMSO-treated synaptosomes whereas bioresmethrin (less toxic 1R-trans stereoisomer) elicited no significant effect (Fig. 2a). Toxic 1R-deltamethrin elicited an average 1.7-fold (±0.03) increase in K+-stimulated Ca2+ influx compared to DMSO-treated synaptosomes at all K+ concentrations whereas the non-toxic 1S-enantiomer of deltamethrin elicited no significant effect (Fig. 2b).

Structure-activity relationships of a cismethrin and b deltamethrin on Ca2+ influx using rat brain synaptosomes. Synaptosomes were treated with 2 × 10−7 M concentrations of pyrethroid in DMSO or DMSO alone. An asterisk indicates that pyrethroid treatment is significantly greater than DMSO treatment (ANOVA, n = 3, Dunnett’s Post Hoc Test, P < 0.05)
Membrane depolarization induced by elevated external K+ evoked endogenous glutamate release only in the presence of external Ca2+ (Fig. 3a, b). Triton X-100 lysed the synaptosomes and the results indicated that not all the glutamate in the synaptosomes was released by 60 K+ (Fig. 3a, b). Total endogenous glutamate conversion was slow due to the low substrate concentration and the high synaptosomal protein concentration in the enzyme-linked assay (see Triton X-100 curve, Fig. 3a).

The effects of cismethrin (2 × 10−7 M) and deltamethrin (2 × 10−7 M) on K+-stimulated glutamate release from rat brain synaptosomes. a Sample time course experiment illustrating that glutamate release is increased by treatment with 60 mM K+ (0 mM K+, non-depolarized). b Glutamate release is dependent on external Ca2+. c The effect of increasing K+ concentrations on deltamethrin-dependent glutamate release. An open triangle indicates that deltamethrin treatment is significantly greater than DMSO treatment (n = 8, unpaired t test, P < 0.05). Triton X-100 (0.5%, X-100)
Deltamethrin (2 × 10−7 M) resulted in an average 1.2-fold (±0.04) increase in glutamate release compared to DMSO-treated synaptosomes that was significant at all K+ concentrations (paired t test, P = 0.017, Fig. 3c). Unlike the Ca2+ influx experiments, only deltamethrin-treated synaptosomes displayed an increased glutamate released while cismethrin (2 × 10−7 M) produced no significant effect (Fig. 3c).
Neither cismethrin nor bioresmethrin produced any effect on K+-stimulated glutamate release (Fig. 4a). 1R-deltamethrin resulted in an average 1.5-fold (±0.06) increase in K+-stimulated glutamate release, which was significantly greater than that caused by DMSO treatment at all K+ concentrations (Fig. 4b). 1R-deltamethrin-dependent glutamate release was also stereospecific since 1S-deltamethrin did not increase release.

Structure-activity relationships of a cismethrin and b deltamethrin on glutamate release from rat brain synaptosomes. Synaptosomes were treated with 2 × 10−7 M concentrations of pyrethroid in DMSO or DMSO alone. An asterisk indicates that pyrethroid treatment is significantly greater than DMSO treatment (ANOVA, n = 3, Dunnett’s Post Hoc Test, P < 0.05)
Both pyrethroids produced concentration-dependent responses on Ca2+ influx (Fig. 5a) but only deltamethrin evoked glutamate release in a concentration-dependent manner (Fig. 5b). ANOVA comparison of the regression lines for the concentration-dependent responses elicited by cismethrin versus deltamethrin in the Ca2+ influx assay were statistically different (F test, P < 0.0001, Fig. 5a). Cismethrin and deltamethrin also resulted in different concentration-dependent responses in the glutamate release assay (Fig. 5b) in that cismethrin failed to elicit a concentration-dependent response.

The effects of increasing concentrations of cismethrin (filled triangle) or deltamethrin (filled square) on K+-stimulated a Ca2+ influx and b endogenous glutamate release using rat brain synaptosomes. KCl (60 mM) was used to depolarize the synaptosomes. Synaptosomes were treated with pyrethroid concentrations from 2 × 10−17 to 2 × 10−5 M (n ≥ 3). Reproduced from Symington et al. 2007a with permission, Copyright 2007
Deltamethrin was several orders of magnitude (∼6.4 × 105) more potent on Ca2+ influx than cismethrin as judged by their EC50 values and only deltamethrin resulted in a significant enhancement in glutamate release (Table 1). Cismethrin was 1.8-fold more efficacious than deltamethrin in the Ca2+ influx assay. The efficacy value of cismethrin is questionable, however, since saturation was not obtained and Ca2+ influx occurred only at high concentrations. Nevertheless, deltamethrin was more efficacious than cismethrin in eliciting Ca2+ influx over the concentration range of 10−12 to 10−7 M, the same range over which deltamethrin also enhanced glutamate release.
These results suggest that the manner in which Ca2+ enters the synaptosomes is different for each of the pyrethroids. To better understand the mechanisms of Ca2+ influx elicited by cismethrin or deltamethrin, selective ion channel blockers were used to examine voltage-sensitive calcium and sodium channels.
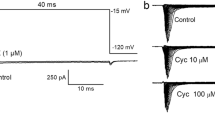
The specific Cav2.2 blocker, ω-conotoxin GVIA (GVIA, 1 μM), elicited a 21% (−1.3-fold) reduction in K+-stimulated Ca2+ influx (Fig. 6a) compared to DMSO-treated synaptosomes. Although Ca2+ influx is reduced, it is not significantly less than DMSO-treated synaptosomes. This outcome is primarily due to the substantial Ca2+ influx that occurs via L-type VSCC during the sustained membrane depolarization in the presence of 60 mM K+. Such an influx is not expected to be sensitive to GVIA nor involved in neurotransmitter release (Symington et al. 2007a). Nevertheless, K+-stimulated Ca2+ influx that was enhanced by cismethrin was not significantly altered by GVIA. Deltamethrin-dependent Ca2+ influx, however, was virtually eliminated by GVIA compared to GVIA + DMSO-treated synaptosomes and was 63% (−2.7-fold) lower than synaptosomes treated with only deltamethrin. Together, these results suggest that deltamethrin-dependent, K+-stimulated Ca2+ influx is occurring primarily via Cav2.2.

The effect of ion channel toxins on pyrethroid-dependent, K+-stimulated a Ca2+ influx and b endogenous glutamate release using rat brain synaptosomes. Synaptosomes were treated with a 2 × 10−7 M concentration of pyrethroid in DMSO or DMSO alone. a The effect of voltage-sensitive calcium channel antagonists; ω-conotoxin GVIA (GVIA, 1 μM) and the voltage-sensitive sodium channel antagonist, tetrodotoxin (TTX, 10 μM) pretreatment on pyrethroid-evoked Ca2+ influx. b The effect of 1 μM GVIA and 10 μM TTX pretreatment on pyrethroid-dependent glutamate release. An asterisk indicates that pyrethroid treatment was significantly different than the corresponding DMSO treated control (n ≥ 3, ANOVA, P < 0.05). A closed diamond indicates that toxin pretreatment is significantly less than the corresponding DMSO treatment (n ≥ 3, ANOVA, P < 0.05). An open triangle indicates that synaptosomes treated with pyrethroid in the presence of the toxin is significantly different than synaptosomes treated with pyrethroid in the absence of the toxin (ANOVA, n ≥ 3, Newman–Keuls Post Hoc Test, P < 0.05)
TTX, a specific voltage-sensitive sodium channel blocker, resulted in no significant change in the amount K+-stimulated Ca2+ influx compared to DMSO-treated synaptosomes although there was a slight increase in the presence of TTX (Fig. 6a). As previously shown in the absence of channel blockers, cismethrin increased K+-stimulated Ca2+ influx by 1.4-fold. In the combined presence of TTX and cismethrin, K+-stimulated Ca2+ influx, however, was significantly reduced by 50% (−2-fold) versus synaptosomes pretreated with TTX + DMSO. TTX pretreatment resulted in no similar reduction in deltamethrin-dependent, K+-stimulated Ca2+ influx, where deltamethrin treatment still averaged a 2.1-fold increase in Ca2+ influx versus TTX + DMSO-treated synaptosomes (Fig. 6a). This value was also higher than the relative fold increase evoked by deltamethrin in the absence of TTX (Fig. 6a, 1.5-fold). Thus, deltamethrin-dependent, K+-stimulated Ca2+ influx was not inhibited by TTX but was increased. This result is consistent, nonetheless, with the K+-stimulated membrane depolarization method used. Voltage-sensitive K+ channels are freely permeable to K+ at resting membrane potential (approx. −70 mV) and the addition of 60 mM K+ externally caused K+ to enter synaptosomes, producing a net membrane potential change of ∼65 mV (Blaustein and Goldring 1975) and depolarizing the membrane to approx. −5 mV. With CNS voltage-sensitive sodium channels blocked by TTX, Na+ conductance does not occur and subsequent K+ conductance is greatly reduced. Thus, the membrane potential stays persistently at −5 mV. At this depolarizing potential, Cav2.2 is operating near its peak current and only ∼60–70% of this current is inhibited by subsequent channel inactivation (Symington et al. 2007b). The slight increase in Ca2+ influx in the presence of TTX compared to its absence is consistent with this scenario. Similarly, deltamethrin resulted in a hyperpolarizing shift in the midpoint potential of activation and slowed inactivation of heterologously expressed Cav2.2 (Symington and Clark 2005). Both actions are consistent with the increased Ca2+ influx that occurred with synaptosomes treated with deltamethrin in the presence of TTX.
Overall, these results suggest that cismethrin is primarily increasing Ca2+ influx via a mechanism that is inhibited by TTX whereas the effect caused by deltamethrin is increased by TTX. Cismethrin-dependent, K+-stimulated Ca2+ influx is likely occurring as consequence of Na+ influx via TTX-sensitive voltage-sensitive sodium channels, followed by Na +i /Ca 2+o exchange. Deltamethrin-dependent, K+-stimulated Ca2+ influx is highly sensitive to GVIA and implies a role for Cav2.2. Thus, cismethrin and deltamethrin may differentially affect voltage-sensitive ion channels at presynaptic nerve terminals during K+-stimulated depolarization.
GVIA (1 μM) resulted in an ∼53% (−2.1-fold) reduction in K+-stimulated glutamate release versus synaptosomes treated with DMSO only, which is consistent with the role of Cav2.2 in neurotransmitter release (Fig. 6b) (Meder et al. 1999). Neither cismethrin nor deltamethrin enhanced K+-stimulated glutamate release from synaptosomes pretreated with GVIA (Fig. 6b). GVIA pretreatment reduced deltamethrin-evoked glutamate release by 70% (−3.3-fold) compared to synaptosomes treated only with deltamethrin (n ≥ 3, ANOVA, P < 0.05).
TTX (1 μM) alone resulted in no significant change in the amount of K+-stimulated glutamate release from DMSO-treated synaptosomes (Fig. 6b), which is consistent with previous results (Meder et al. 1999). Cismethrin resulted a 36.2% (−1.6-fold) reduction in glutamate release in the presence of TTX (Fig. 6b) and is consistent with the results obtained in the Ca2+ influx assay with cismethrin (Fig. 5a). Deltamethrin resulted in a 1.4-fold increase of glutamate released in the presence of TTX (Fig. 6b), a value similar to that obtained in the absence of TTX (1.4-fold) (Fig. 6b).
Collectively, these results show that deltamethrin-dependent, K+-staimulated Ca2+ influx and subsequent glutamate release are not occurring as a consequence of TTX-sensitive voltage-sensitive sodium channel modification and suggests additional sites of action at presynaptic nerve terminals, most likely Cav2.2.
Action of deltamethrin on heterologously expressed Cav2.2
Sample current traces indicate a stereospecific action of the 1R versus 1S enantiomers of deltamethrin (10−7 M) on wild type Cav2.2 (Fig. 7a). 1R-deltamethrin reduced Ba2+ peak current whereas 1S-deltamethrin had no effect on Ba2+ peak current (Fig. 7a). Using current-voltage relationships, treatment with 1R-deltamethrin decreased the average peak current value to 0.53 ± 0.10 compared to the normalized control (value of untreated control normalized to 1) whereas treatment with 1S-deltamethrin was without effect (0.97 ± 0.05) (t test, P < 0.05, Fig. 8a).

Representative current recordings illustrating the effects of 1R- or 1S-deltamethrin (1 × 10−7 M) on wild type Cav2.2 α1B-subunit (a) and T422E mutant (b) co-expressed with the β3-subunit in Xenopus oocytes under steady-state depolarization. Currents were evoked by a step depolarization to 0 mV from a holding potential of −80 mV for 2.5 s. All recordings were made using 5 mM Ba2+ as the charge carrier (n > 5)

Effect of toxic (1R-) and nontoxic (1S-) deltamethrin (1 × 10−7 M) on the current-voltage relationships of wild type Cav2.2 α1B-subunit (a) and the T422E mutant (b) co-expressed with β3-subunit in Xenopus oocytes. Currents were evoked with 5 mV step depolarizations for 150 ms each from a holding potential of −80 mV to +40 mV. All recordings were made using 5 mM Ba2+ as the charge carrier (n > 5)
Conversely, 1R-deltamethrin increased Ba2+ peak current when the T422E Cav2.2 was similarly examined and 1S-deltamethrin was substantially less effective in increasing Ba2+ current (note scale difference between traces, Fig. 7b). Using current-voltage relationships, the normalized average peak currents for 1R- and 1S-deltamethrin were 1.49 ± 0.12 and 0.94 ± 0.12, respectively (t test, P < 0.05, Fig. 8b). These results establish a stereospecific action for deltamethrin on the T422E Cav2.2 that is opposite from that obtained with the wild type (T422) channel.
Discussion
Our current findings establish a stereospecific and agonistic action of cismethrin and deltamethrin on Ca2+ influx into rat brain synaptosomes depolarized by K+. The data also suggest that the manner in which Ca2+ enters presynaptic nerve terminals occurs by different mechanisms since only deltamethrin-dependent Ca2+ influx results in increased glutamate release.
Previous electrophysiological findings showed that deltamethrin directly modified the function of heterologously expressed Cav2.2 by causing a prolongation of the activation and inactivation kinetics, a hyperpolarizing shift in the midpoint potential of activation, and a reduction of Ba2+ peak current (Symington and Clark 2005). These results support the claim that deltamethrin, and possibly other CS-syndrome pyrethroids, target Cav2.2 associated with the presynaptic nerve terminal in the CNS (Clark and Brooks 1989a).
Our current electrophysiological experiments conducted with T422E Cav2.2 support the contention that phosphorylation is involved with the action of deltamethrin, a process that significantly increased deltamethrin-dependent Ba2+ peak current and mimics the effect of deltamethrin in vivo. Other support of this contention is provided by the observation that protein phosphorylation patterns in synaptosomal preparations are modified by deltamethrin (Matsumura et al. 1989), possibly by a PKC-dependent pathway (Enan and Matsumura 1993). PKC is the protein kinase that phosphorylates the T422 residue in Cav2.2, which increases calcium channel activity (Zamponi et al. 1997).
Our biochemical results on Ca2+ influx and endogenous glutamate release using functional synaptosomes, along with other in situ electrophysiological measurements conducted with deltamethrin (Symington et al. 1999b; Duce et al. 1999b), substantiates that deltamethrin acts as selective VSCC modulator. Moreover, the effect of deltamethrin on heterologously expressed Cav2.2 is substantially different from those results obtained when heterologously expressed calcium channels (Cav1, Cav2.1 and Cav2.3) were treated with bioallethrin (Hildebrand et al. 2004). These results support the contention that effects of pyrethroids on VSCC are analog-specific, isoform-specific, and may differ under in vivo versus in vitro conditions where the in vivo response is governed by the collective regulation of all the proteins and co-factors localized to the specific channel being investigated.
In summary, our functional biochemical assays yielded results that support our hypothesis that the CS-syndrome pyrethroids are VSCC agonists, resulting in increased Ca2+ influx and glutamate release under K+-stimulated depolarizing conditions. The glutamate release data establish distinctly different biochemical profiles for cismethrin versus deltamethrin at rat presynaptic nerve terminals that are consistent with previous reported in vivo acute neurotoxic responses during the onset of the T- versus CS-syndromes (Aldridge et al. 1978; Hossain et al 2004). Furthermore, the reduction of deltamethrin-dependent Ca2+ influx and glutamate release by GVIA and the specific inhibition of cismethrin-dependent Ca2+ influx by TTX provide additional evidence that supports distinct biochemical profiles for these two pyrethroids at presynaptic nerve terminals. Lastly, the involvement of post-translational modifications on the function of Cav2.2 is well documented but not completely understood. The opposite effect of deltamethrin on peak current with wild type versus T422E Cav2.2 establishes a need for additional research on the role of channel phosphorylation in the action of deltamethrin on the gating kinetics of ion channel targets.
References
Aldridge WN, Clothier B, Froshaw P, Johnson MK, Parker VH, Price RJ, Skilleter DN, Verscholyle RD, Stevens C (1978) The effect of DDT and the pyrethroids cismethrin and decamethrin on the acetyl choline and cyclic nucleotide content of rat brain. Biochem Pharmacol 27:1703–1706
Blaustein MP, Goldring JM (1975) Membrane potentials in pinched-off presynaptic nerve terminals monitored with fluorescent probe: Evidence that synaptosomes have potassium diffusion potentials. J Physiol 247:589–615
Bloomquist JR, Soderlund DM (1988) Pyrethroid insecticides and DDT modify alkaloid-dependent sodium channel activation and its enhancement by sea anemone toxin. Mol Pharmacol 33:543–550
Brooks MW, Clark JM (1987) Enhancement of norepinephrine release from rat brain synaptosomes by alpha-cyano pyrethroids. Pestic Biochem Physiol 28:127–139
Catterall WA (1997) Modulation of sodium and calcium channels by protein phosphorylation and G proteins. Adv Second Messenger Phosphoprotein Res 31:159–181
Catterall WA (1998) Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium 24:307–323
Catterall WA (1999) Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann N Y Acad Sci 868:144–159
Clark JM (1994) Effects and mechanisms of action of pyrethrin and pyrethroid insecticides. In: LWCaRS Dyer (eds) Handbook of neurotoxicology. Marcel Dekker Inc., New York, Basel, Hong Kong, pp 511–46
Clark JM, Brooks MW (1989a) Neurotoxicology of pyrethroids: single or multiple mechanisms of action? J Environ Toxicol Chem 8:361–372
Clark JM, Brooks MW (1989b) Role of ion channels and intraterminal calcium homeostasis in the action of deltamethrin at presynaptic nerve terminals. Biochem Pharmacol 38:2233–2245
Clark JM, Marion JR (1990) Enhanced neurotransmitter release by pyrethroid insecticides. In: Narahashi T, Chambers JE, Chambers H (eds) Insecticidal action: from molecule to organism. Plenum Press, New York NY, pp 139–168
Clark JM, Edman SJ, Nagy SR, Conhoto A, Hecht F, Van Houten J (1995) Action of DDT and pyrethroids on the calcium channel in Paramecium tetraurelia. In: Clark JM (eds) Molecular action of insecticides on ion channels. American Chemical Society, Washington DC, pp 173–190
Cooper CB, Arnot MI, Feng ZP, Jarvis SE, Hamid J, Zamponi GW (2000) Cross-talk between G-protein and protein kinase C modulation of N-type calcium channels is dependent on the G-protein beta subunit isoform. J Biol Chem 275:40777–40781
De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP (1997) Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature 385:446–450
Doherty JD, Nishimura K, Kurihara N, Fujita T (1987) Promotion of norepinephrine released and inhibition of calcium uptake by pyrethroids in rat brain synaptosomes. Pestic Biochem Physiol 29:187–196
Duce I, Warburton S, Khan T, Thompson A, Green C (1999a) Insect calcium channels. In Neurotox 98. SCI Oxford, UK
Duce IR, Khan TR, Green AC, Thompson AJ, Warburton SPM, Wong J (1999b) Calcium channels in insects. In: Beadle DJ (ed) Progress in Neuropharmacology and neurotoxicology of pesticides and drugs. The Royal Society of Chemistry Oxford, UK pp 56–66
Eells JT, Dubocovich ML (1988) Pyrethroid insecticides evoke neurotransmitter release from rabbit striatal slices. J Pharmacol Exp Ther 246:514–521
Ehrlich BE, Jacobson AR, Hinrichsen R, Sayre LM, Forte MA (1988) Paramecium calcium channels are blocked by a family of calmodulin antagonists. Proc Natl Acad Sci USA 85:5718–5722
Enan E, Matsumura F (1991) Stimulation of protein phosphorylation in intact rat brain synaptosomes by a pyrethroid insecticide, deltamethrin. Pestic Biochem Physiol 39:182–195
Enan E, Matsumura F (1993) Activation of phosphoinositide/protein kinase C pathway in rat brain tissue by pyrethroids. Biochem Pharmacol 45:703–710
Fink K, Meder WP, Clusmann H, Gothert M (2002) Ca2+ entry via P/Q-type Ca2+ channels and the Na+/Ca2+ exchanger in rat and human neocortical synaptosomes. Naunyn Schmiedebergs Arch Pharmacol 366:458–463
Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260:3440–3450
Guo-lei F, Marion JR, Clark JM (1992) Suppression of pyrethroid-dependent neurotransmitter release from synaptosomes of knockdown-resistant house flies under pulsed depolarization conditions during continuous perfusion. Pestic Biochem Physiol 42:64–77
Hamid J, Nelson D, Spaetgens R, Dubel SJ, Snutch TP, Zamponi GW (1999) Identification of an integration center for cross-talk between protein kinase C and G protein modulation of N-type calcium channels. J Biol Chem 274:6195–6202
Harlow ML, Ress D, Stoschek A, Marshall RM, McMahan UJ (2001) The architecture of active zone material at the frog’s neuromuscular junction. Nature 409:479–484
Hildebrand ME, McRory JE, Snutch TP, Stea A (2004) Mammalian voltage-gated calcium channels are potently blocked by the pyrethroid insecticide allethrin. J Pharmacol Exp Ther 308:805–813
Hossain MM, Suzuki T, Sato I, Takewaki T, Suzuki K, Kobayashi H (2004) The modulatory effect of pyrethroids on acetylcholine release in the hippocampus of freely moving rats. Neurotoxicology 25:825–833
Iredale PA, Dickenson JM (1995) Measurement of intracellular free calcium ion concentration in cell populations using fura-2. Methods Mol Biol 41:203–213
Ishikawa Y, Charalambous P, Matsumura F (1989) Modification by pyrethroids and DDT of phosphorylation activities of rat brain sodium channel. Biochem Pharmacol 38:2449–2457
Kanemoto Y, Enan EE, Matsumura F, Miyazawa M (1992) Time-dependent changes in protein phosphorylation patterns in rat brain synaptosomes caused by deltamethrin. Pestic Sci 34:281–290
Katz B, Miledi R (1967) The release of acetylcholine from nerve endings by graded electric pulses. Proc R Soc Lond B Biol Sci 167:23–38
Koenig JH, Yamaoka K, Ikeda K (1998) Omega images at the active zone may be endocytotic rather than exocytotic: implications for the vesicle hypothesis of transmitter release. Proc Natl Acad Sci USA 95:12677–12682
Li D, Wang F, Lai M, Chen Y, Zhang JF (2005) A protein phosphatase 2calpha-Ca2+ channel complex for dephosphorylation of neuronal Ca2+ channels phosphorylated by protein kinase C. J Neurosci 25:1914–1923
Lin Z, Haus S, Edgerton J, Lipscombe D (1997) Identification of functionally distinct isoforms of the N-type Ca2+ channel in rat sympathetic ganglia and brain. Neuron 18:153–166
Matsumura F, Clark JM, Matsumura FM (1989) Deltamethrin causes changes in protein phosphorylation activities associated with post-depolarization events in the synaptosomes from the optic lobe of squid, Loligo pealei. Comp Biochem Physiol C 94:381–390
Meder W, Fink K, Zentner J, Gothert M (1999) Calcium channels involved in K+- and veratridine-induced increase of cytosolic calcium concentration in human cerebral cortical synaptosomes. J Pharmacol Exp Ther 290:1126–1131
Miyazawa M, Matsumura F (1990) Effects of deltamethrin on protein phosphorylation and dephosphorylation process in the nerve fibers of the American lobster, Homarus americanus L. Pestic Biochem and Physiol 32:147–155
Narahashi T (1992) Nerve membrane Na+ channels as targets of insecticides. Trends Pharmacol Sci 13:236–241
Narahashi T, Tsunoo A, Yoshii M (1987) Characterization of two types of calcium channels in mouse neuroblastoma cells. J Physiol (Lond) 383:231–249
Nicholls DG, Sihra TS, Sanchez-Prieto J (1987) Calcium-dependent and -independent release of glutamate from synaptosomes monitored by continuous fluorometry. J Neurochem 49:50–57
Nichols RA, Haycock JW, Wang JK, Greengard P (1987) Phorbol ester enhancement of neurotransmitter release from rat brain synaptosomes. J Neurochem 48:615–621
Nicholson RA, Wilson RC, Potter C, Black MH (1987) Pyrethroid- and DDT-evoked release of GABA from the nervous system in vitro. In: Miyamoto J, Kearney PC (eds) Pesticide chemistry: human welfare and the environment, Pergamon. Oxford, UK, pp 75–78
Osborne MP, Pepper DR, Hein PJD (1995) Site-insensitive mechanisms in knockdown resistant strains of housefly larva, Musca domestica In: Clark JM (eds) Molecular action of insecticides on ion channels. American Chemical Society, Washington DC, pp 128–148
Rossie S (1999) Regulation of voltage-sensitive sodium and calcium channels by phosphorylation. In: Armstrong DL, Rossie S (eds) Advances in second messenger and phosphoprotein research. Academic, San Diego, pp 23–48
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85
Soderlund DM, Bloomquist JR (1989) Neurotoxic actions of pyrethroid insecticides. Ann Rev Entomol 34:77–96
Soderlund DM, Clark JM, Sheets LP, Mullin LS, Piccirillo VJ, Sargent D, Stevens JT, Weiner ML (2002) Mechanisms of pyrethroid neurotoxicity: implications for cumulative risk assessment. Toxicology 171:3–59
Soreq H, Seidman S (1992) Xenopus oocyte microinjection: from gene to protein. Methods Enzymol 207:225–265
Stea A, Soong TW, Snutch TP (1995) Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron 15:929–940
Symington SB, Clark JM (2005) Action of deltamethrin on voltage-sensitive calcium channels in rat brain. Pestic Biochem Physiol 82:1–15
Symington SB, Zhang A, Clark JM (1999a) The action of pyrethroids on the voltage-sensitive calcium channel of Paramecium tetraurelia. Pestic Sci 55:1035–1037
Symington SB, Zhang A, Karstens W, Van Houten J, Clark JM (1999b) Characterization of pyrethroid action on ciliary calcium channels in Paramecium tetraurelia. Pestic Biochem Physiol 65:181–193
Symington SB, Frisbie RK, Lu KD, Clark JM (2007a) Action of cismethrin and deltamethrin on functional attributes of isolated presynaptic nerve terminals from rat brain. Pestic Biochem Physiol (in press)
Symington SB, Frisbie RK, Kim H-J, Clark JM (2007b) Mutation of threonine 422 to glutamic acid mimics the phosphorylation state and alters the action of deltamethrin on Cav2.2. Pestic Biochem Physiol (in press)
Trainer VL, McPhee JC, Boutelet-Bochan H, Baker C, Scheuer T, Babin D, Demoute JP, Guedin D, Catterall WA (1997) High affinity binding of pyrethroids to the alpha subunit of brain sodium channels. Mol Pharmacol 51:651–657
Turner TJ, Adams ME, Dunlap K (1993) Multiple Ca2+ channel types coexist to regulate synaptosomal neurotransmitter release. Proc Natl Acad Sci USA 90:9518–9522
Wennemuth G, Westenbroek RE, Xu T, Hille B, Babcock DF (2000) CaV2.2 and CaV2.3 (N- and R-type) Ca2+ channels in depolarization-evoked entry of Ca2+ into mouse sperm. J Biol Chem 275:21210–21217
Yoshii M, Tsunoo A, Narahashi T (1985) Effects of pyrethroids and veratridine on two types of calcium channels in neuroblastoma cells. Presented at Society for Neuroscience, Dallas
Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP (1997) Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature 385:442–446
Zhang A. (1996) Presynaptic actions of insecticidal dihydropyrazoles in mammalian brain. Dissertation thesis. Simon Frazier University, Vancouver. pp 139
Zhang A, Nicholson RA (1994) RH-3421, a potent dihydropyrazole insecticide, inhibits depolarization- stimulated rises in free [Ca2+] and 45Ca2+ uptake in mammalian synaptosomes. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol 108:307–310
Acknowledgments
We thank Dr. Diana Lipscombe (Brown University, Providence, RI) for providing us the Cav2.2 subunits (α−1B-d and β3). We thank the Pyrethroid Working Group (PWG: Adventis CropSci., Bayer Corp., DuPont Ag. Prod., FMC Corp., Valent USA Corp., and Syngenta) for providing the pyrethroids used in this study. Partial support For S. Symington was provided by RI-INBRE (NIH/NCRR. #P20PR016457).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Clark, J.M., Symington, S.B. Pyrethroid action on calcium channels: neurotoxicological implications. Invert Neurosci 7, 3–16 (2007). https://doi.org/10.1007/s10158-006-0038-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10158-006-0038-7