
Abstract
The dopamine receptor type 1 (D1R) and the dopamine receptor type 5 (D5R), which are often grouped as D1R-like due to their sequence and signaling similarities, exhibit high levels of constitutive activity. The molecular basis for this agonist-independent activation has been well characterized through biochemical and mutagenesis in vitro studies. In this regard, it was reported that many antipsychotic drugs act as inverse agonists of D1R-like constitutive activity. On the other hand, D1R is highly expressed in the medial prefrontal cortex (mPFC), a brain area with important functions such as working memory. Here, we studied the impact of D1R-like constitutive activity and chlorpromazine (CPZ), an antipsychotic drug and D1R-like inverse agonist, on various neuronal CaV conductances, and we explored its effect on calcium-dependent neuronal functions in the mouse medial mPFC. Using ex vivo brain slices containing the mPFC and transfected HEK293T cells, we found that CPZ reduces CaV2.2 currents by occluding D1R-like constitutive activity, in agreement with a mechanism previously reported by our lab, whereas CPZ directly inhibits CaV1 currents in a D1R-like activity independent manner. In contrast, CPZ and D1R constitutive activity did not affect CaV2.1, CaV2.3, or CaV3 currents. Finally, we found that CPZ reduces excitatory postsynaptic responses in mPFC neurons. Our results contribute to understanding CPZ molecular targets in neurons and describe a novel physiological consequence of CPZ non-canonical action as a D1R-like inverse agonist in the mouse brain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The dopaminergic system controls many cognitive, motor, emotional, and social functions that ultimately regulate behavior [1, 2]. Dopamine acts via dopamine receptors (D1-5R), a family of G-protein-coupled receptors (GPCR) that are highly expressed throughout the mammalian brain [3]. Based on their similarities of sequence and their signaling properties, dopamine receptors can be grouped in two classes (i.e., D1R-like − D1R and D5R- and D2R-like − D2R, D3R, and D4R-) [4]. In particular, D1R receptors are highly expressed in the medial prefrontal cortex (mPFC), where their activity is crucial for cognitive processes, such as working memory, in both physiological and pathological states [5]. Notably, D1R-like receptors exhibit high levels of constitutive activity, an agonist-independent and active conformational state that produces signaling in the absence of dopamine [6]. Thus, D1R-like receptors show both agonist-dependent and agonist-independent active modes. Years of research have identified numerous cellular targets for dopamine receptors ligand-evoked activation, many of them linked to a specific behavior in the healthy and the pathological brain [7,8,9]. In contrast, while the molecular basis for the basal activity of D1R-like has been well characterized in biochemical and mutagenesis in vitro studies [6, 10,11,12], few have explored its relevance in physiological contexts [13,14,15,16]. In this work, we studied the effects of constitutive D1R-like activity on native voltage-gated calcium channels (CaV), proteins that are knowingly sensitive to GPCR activity and are key for neuronal function.
Since the first evidence for GPCR constitutive activity was obtained for the δ-opioid receptor [17], more than 60 wild-type GPCRs have been confirmed to exhibit constitutive activity [18]. Many of these constitutively active GPCRs are receptors for neurotransmitters, and it was proposed that their basal signaling provides a tonic support for basal neuronal activity [19]. Studying the physiological impact of D1R-like and other GPCR constitutive activity can be challenging due to the presence of endogenous ligands that simultaneously trigger agonist-evoked activity. In this regard, the use of ligands that reduce constitutive signaling—inverse agonists—to pharmacologically manipulate the level of constitutive activity in both native and recombinant systems has been a crucial strategy in the characterization of GPCR constitutive activity and its relevance in vivo [20,21,22]. Inverse agonists have been reported for the majority (~ 75%) of constitutively active GPCRs [19], and at least two of them are endogenous: the Agouti-related peptide (AgRP) for the melanocortin-4-receptor (MC4R) and the liver-expressed antimicrobial peptide 2 (LEAP-2) for the ghrelin receptor (GHSR) [23, 24]. However, many of these ligands are synthetic and some are commonly used drugs with a high pharmacotherapeutic relevance in the treatment of depression and schizophrenia [19]. In particular, several antipsychotic drugs, classically described as D2R antagonists, were proven through in vitro studies to act as inverse agonists of D1R-like constitutive activity [10]. Among these, chlorpromazine (CPZ) is currently one of the most frequently used antipsychotic drugs and it is known to display D1R-like inverse agonist properties [25], among other cellular targets [26]. The extent to which such D1R-like inverse agonism contributes to the clinical pharmacology of CPZ remains unknown.
Neurons simultaneously express many CaV subtypes with different sublocalizations and biophysical properties that determine their involvement in specific neuronal functions [27]. The activation of neuronal CaV is one of the main sources of activity-dependent calcium entry that contributes to firing, initiates synaptic plasticity mechanisms and modulates transcription [28]. One important feature that shapes CaV function is the voltage range at which they are activated, and in this regard, their currents can be grouped into two categories: high voltage-activated (HVA) and low voltage-activated (LVA) [29]. The CaV1 and CaV2 subfamilies mainly contribute to HVA currents, while the CaV3 subfamily supports LVA currents. We have shown that certain CaV subtypes display a particularly high sensitivity to different GPCRs constitutive signaling, including D1R-like [14, 20, 30,31,32]. Specifically, previous work from our lab and others showed that D1R-like constitutive activity upregulates CaV2.2 currents in mPFC layer 5/6 pyramidal neurons, increasing channel density in postsynaptic membranes [14, 33]. In this context, it would be significant to determine if D1R-like constitutive activation and an inverse agonist currently used in clinical treatments, such as CPZ, affect other CaV subtypes expressed in these mPFC neurons.
In this study, we investigated the effects of a D1R-like inverse agonist, CPZ, on a variety of CaV subtypes expressed in the mouse mPFC. We combined in vivo administration of CPZ with ex vivo electrophysiological recordings of native CaV currents from mPFC layer 5/6 pyramidal neurons. We dissected the effect of CPZ on the different CaV subtypes that contribute to the total native current in these neurons and, specifically, explored the role of D1R-like constitutive activity in a controlled heterologous expression system. Finally, we explored the impact of CPZ on excitatory postsynaptic activity and transcriptional activity as possible outcomes of somatic CaV current modulation.
Materials and Methods
Animals
This study was performed using 4- to 8-week-old wild-type (WT) C57BL/6 mice (Mus musculus) of both sexes counterbalanced (n = 75). Mice were bred and housed at the Multidisciplinary Institute of Cell Biology (IMBICE) animal facility with a 12-h light/dark cycle, controlled room temperature (22 °C ± 2 °C) and ad libitum access to food (Gepsa Pilar Group AS, Argentina) and water. All experiments in this study received approval from the Institutional Animal Care and Use Committee of the IMBICE (approval ID 120,319), in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Research Council (US).
Drugs
Chlorpromazine hydrochloride (CPZ, 25 mg/ml injectable blister, #50–53-3; Duncan Laboratory AS, Argentina) was donated by Dr. Martinez Mónaco and Dr. Pinedo (Italian Hospital of La Plata, Buenos Aires, Argentina) and stored at 4° C, protected from light. Chlorpromazine working solutions were prepared fresh on experimental days, using sterile milli-Q water (for preincubations of transfected HEK293T cells), artificial cerebrospinal fluid (aCSF (detailed composition below), for intra-mPFC injections in mice), or sterile saline (0.9% NaCl, for intraperitoneal (IP) injections in mice). Additionally, the following drugs were used as indicated for each experiment: the sodium channel blocker tetrodotoxin (TTX, #4368–28-9, Sigma-Aldrich, USA), the sodium channel blocker lidocaine N-ethyl bromide (QX-314, #552,233, Calbiochem, USA), the CaV1 blocker nifedipine (#N-7634, Sigma-Aldrich, USA), the CaV2.1 blocker ω-agatoxin IVA (#145,017–83-0, Alomone Labs, Israel), the CaV2.2 blocker ω-conotoxin GVIA (#106,375–28-4, Alomone Labs, Israel), the CaV2.3 blocker SNX-482 (#203,460–30-4, Peptide Institute Inc., Japan), the CaV3 blocker TTA-P2 (#T-155, Alomone Labs, Israel), nickel (II) chloride hexahydrate (NiCl2, #2,000,979,900, Biopack Analytical Systems AS, Argentina), dopamine hydrochloride (#62–31-7, Sigma-Aldrich, USA), the D1R-like antagonist R( +)-SCH-23390 (#125,941–87-9, Sigma-Aldrich, USA), and the D1R-like agonist ( ±)-SKF-38393 hydrochloride (#62,717–42-4, Sigma-Aldrich, USA).
Animal Treatment
Stereotaxic surgeries were performed in 8-week-old WT mice in order to place a single guide cannula (22G, P1 Technologies, USA) at the mPFC (placement coordinates AP: + 1.9, ML: + 0.5, and DV: − 2.0 mm, according to the Mouse Brain Atlas of Paxinos and Franklin [34]). All surgeries were performed in the surgical room of the IMBICE animal facility in an aseptic environment as required. Mice were anaesthetized with Ketamine (150 mg/kg) and Xylazine (15 mg/kg). Flunixin meglumine (Flumeg 5%) was used as an analgesic. After surgery, mice were allowed to recover for 5 days with daily manipulations of the cannula to minimize stress. Twenty-four hours before the experimental day, mice were intra-mPFC injected with 0.5 µl of vehicle alone (aCSF, detailed composition below) or containing CPZ (1 µg/mouse) or SCH-23390 (1 µg/mouse). Additionally, mice were treated by IP injections (0.1 ml/10 g) with vehicle alone (sterile 0.9% NaCl) or containing CPZ (1 mg/kg) or SKF-38393 (3 mg/kg) at 24 h, 2 h, and/or 1 h before sacrifice, as indicated for each data set.
Brain Slice Preparations for Electrophysiology
Four- to 8-week-old male and female WT mice were anaesthetized with isoflurane (2%) and immediately decapitated. Brains were quickly removed and immersed in ice-cold 95% O2 and 5% CO2-equilibrated cutting solution containing (in mM) 110 choline chloride, 25 glucose, 25 NaHCO3, 7 MgCl2, 11.6 ascorbic acid, 3.1 sodium pyruvate, 2.5 KCl, 1.25 NaH2PO4, and 0.5 CaCl2, pH 7.4. Coronal brain slices including the mPFC (∼300 μM, 1.5–2.5 mm anterior to bregma) were obtained using a vibratory tissue slicer (PELCO easiSlicer, #11,000, Ted Pella Inc., USA) and then transferred to an incubation chamber filled with 95% O2, 5%CO2–equilibrated aCSF containing (in mM) 124 NaCl, 26.2 NaHCO3, 11 glucose, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, and 1 NaH2PO4, pH 7.4. Slices were maintained at 37 °C for 15 min and left to recover at room temperature (∼23 °C) for 30 min before recordings.
Brain Slice Preparations for Immunohistochemistry
Eight-week-old male WT mice were individually housed 3 days before the experimental day and daily handled in order to minimize stress. Twenty-four hours before sacrifice, mice were IP injected with vehicle alone or containing CPZ (1 mg/kg). On the experimental day, mice were IP injected with vehicle alone or containing SKF-38393 (3 mg/kg) and transcardially perfused with formalin 2 h after treatment. Mice brains were removed, postfixed in formalin for 2 h, and cryopreserved in 20% sucrose overnight. Then, brains were frozen and 4 series of coronal 40 µm slices were obtained with a cryostat (Leica CM1520, Germany). One series of coronal sections was used to perform an anti-c-Fos immunohistochemistry as previously described [35]. Briefly, brain sections were washed with phosphate-buffered saline (PBS), incubated with 0.5% H2O2 for 30 min, incubated with blocking solution (2% adult horse serum and 0.25% Triton X-100 in PBS) for 1 h, and then incubated with an anti-c-Fos antibody (1:20,000, #PC38-100UL; Oncogene, USA) for 48 h at 4 °C. Afterwards, brain sections were sequentially incubated with a biotinylated anti-rabbit antibody (1:3,000, #711–065-152; Jackson ImmunoResearch Laboratories, USA), with avidin–biotin complex (Vectastain Elite Kit, #PK-6100), and the reaction was revealed with 2′,2′-diaminobencidine (DAB)/NiCl2 solution in order to generate a black/purple precipitate in c-Fos-immunoreactive (IR) cell nuclei. Brain slices were mounted on glass slides with mounting media (Canada balsam) and bright-field microphotographs were obtained with a Nikon Eclipse 50i microscope equipped with a Nikon DS-Ri1 camera in order to quantify c-For-IR cells in the mPFC. c-Fos-IR cells were quantified using the software Fiji (ImageJ) and expressed as c-Fos-IR cells per hemi-mPFC subdivision.
Cell Culture and Transfections
Human embryonic kidney 293 T (HEK293T) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; #D5030, Gibco, Thermo-Fisher, USA) with 10% FBS (Internegocios AS, Argentina) and subcultured when 80% confluence was reached. For patch clamp experiments, HEK293T cells were cotransfected with plasmids containing human D1R (DRD1, GenBank Acc. No. X55760), voltage-gated calcium channel alpha-1 subunits CaV1.2 (Cacna1c; GB Acc. No. AY728090), CaV1.3 (Cacna1d; GB Acc. No. AF370009), CaV2.1 (Cacna1a; GB Acc. No. AY714490), CaV2.2 (Cacna1b; GB Acc. No. AF055477), CaV3.1 (Cacna1g; GB Acc. No. AF190860), CaV3.2-GFP (Cacna1h; GB Acc. No. NM021098), and CaV3.3 (Cacna1i; GB Acc. No. NM021096) and auxiliary subunits CaVβ3 (Cacnb3; GenBank Acc. No. M88751) and CaVα2δ1 (Cacna2d1; GenBank Acc. No. AF286488), as indicated for each experiment. The D1R (#DRD0100000, cDNA Resource Center, USA) and CaV3.1 (#45,811, AddGene, USA) clones were purchased. The clones for CaV1.2, CaV1.3, CaV2.1, CaV2.2, CaV3.3, CaVβ3, and CaVα2δ1 were kindly donated by Dr. Diane Lipscombe (Brown University, USA), and the CaV3.2-GFP clone was provided by Dr. Emmanuel Bourinet (Université de Montpellier, France).
For electrophysiology, cells were cotransfected with a D1R:CaV molar ratio of 0.1 (equal to 23 ng of D1R per well). Transient transfections were conducted using Lipofectamine 2000 (#11,668,019, Invitrogen, Thermo-Fisher, USA), with the addition of an enhanced GFP (eGFP) containing plasmid to identify transfected cells and the empty plasmid pcDNA3.1 ( +) to complete the total cDNA amount in the transfection mix. Transfected HEK293T cells were kept in culture for 24 h to allow CaV2.X expression or for 48 h to allow CaV1.X and CaV3.X expression. On the day of the experiment, cells were dispersed with 0.25 mg/ml trypsin (#15,090–046) (Gibco, Thermo-Fisher, USA), rinsed twice, and kept in DMEM at room temperature (∼23 °C) during patch clamp experiments.
Additionally, stable and transient transfections were performed for fluorescence resonance energy transfer (FRET) time course of intracellular cyclic adenosine monophosphate (cAMP) experiments. The mTurquoise2-EPAC-cp173Venus-Venus (Epac-S H187) construct was provided by Dr. Kees Jalink (Netherlands Cancer Institute, the Netherlands) [36]. Stable HEK293T-expressing pcDNA3.1/Zeo(1)-mTurquoise2-EPAC-cp173Venus-Venus (Epac-S H187) (HEKT Epac-S H187) cells were obtained by transfection of HEK293T using the K2 Transfection System (Biontex, Germany). After 24 h, cells were seeded in the presence of 25 μg/ml Zeocin (InvivoGen, USA) for 2 weeks and clonal selection was performed in 96-well plates for 2 weeks. Clones were tested for Epac-S H187 by fluorescence spectra (450–650 nm) measurements in a FlexStation 3 Multi-Mode Microplate Reader (Molecular Devices, USA) with excitation at 430 nm. The HEKT Epac-S H187 clone with higher fluorescence emission was chosen for further experiments. The stable clone was grown in DMEM medium supplemented with 10% FBS, 50 μg/ml gentamicin, and 12.5 μg/ml Zeocin. For transient transfections, HEKT Epac-S H187 cells were grown to 80–90% confluency. Plasmids containing the D1R or pcDNA3.1 ( +) cDNA constructs were transfected into cells using the K2 Transfection System (Biontex, Germany), as recommended by the supplier. Assays were performed 48 h after transfection.
Electrophysiology
Brain Slice Electrophysiology
After recovery, the acute coronal mouse brain slices containing the mPFC were transferred to the recording chamber and visualized with an upright Zeiss Examiner A1 microscope (#491,404–0001-000, Zeiss, Germany), a digital camera (Rolera Bolt Scientific CMOS; QImaging, Canada) and Micro-Manager 1.4 open-source microscopy software (Vale Lab, University of California, USA). Layer 5/6 pyramidal neurons of the prelimbic and infralimbic areas of the mPFC were identified by localization and morphology [37]. Patch clamp recordings in brain slices were made with an EPC7 amplifier (HEKA Electronik, Germany) and data were sampled at 20 kHz and filtered at 10 kHz (− 3 dB) using PatchMaster software (HEKA Electronik, Germany). Series resistances of less than 6 MΩ were admitted and compensated 80% with a 10 μs lag time. Leak current was subtracted online using a P/ − 4 protocol, and recordings with leak currents over 100 pA at holding potential were discarded. Whole-cell patch clamp recordings in voltage-clamp mode were conducted at room temperature (∼23 °C) in the previously described aCSF under a continuous flow rate of 2.5 ml/min. The datasets for each experimental condition were obtained from at least three animals on independent experimental days.
To record native calcium currents, recording electrodes with resistances between 4 and 8 MΩ were filled with an internal solution containing (in mM): 115 Cs-methanesulfonate, 20 tetraethylammonium chloride, 10 CsCl, 5 NaCl, 10 HEPES, 10 EGTA, 4 Mg-ATP, and 0.3 Na-GTP, pH 7.4. TTX (1 μM) was added to normal aCSF to block native voltage-gated sodium channels. Neurons were held at resting potential (− 80 mV) and native calcium currents were evoked applying square pulses from holding to 0 mV (100 ms duration) or to − 30 mV (200 ms duration), with a subsequent step at − 60 mV (50 ms duration) before returning to resting potential. Finally, to identify the contribution of the different CaV subtypes to total native calcium currents, the specific blockers nifedipine (10 μM, CaV1 blocker), ω-agatoxin-IVA (1 μM, CaV2.1 blocker), ω-conotoxin-GVIA (1 μM, CaV2.2 blocker), SNX-482 (100 nM, CaV2.3 blocker), NiCl2 (100 μM, CaV2.3 and CaV3 blocker [38, 39]) or TTAP-2 (1 µM, CaV3 blocker) were added to the bath solution.
To record excitatory postsynaptic currents (EPSC), recording electrodes were filled with an internal solution containing (in mM): 115 Cs-methanesulfonate, 20 tetraethylammonium chloride, 5 NaCl, 10 HEPES, 10 EGTA, 4 Mg- ATP, and 0.3 Na-GTP, pH 7.4. Cells were held a − 70 mV as resting potential. For the electrically evoked EPSC from layer 5/6 pyramidal neurons, QX-314 (10 mM) was added to the internal solution to avoid action potentials in the recorded neuron and a stimulating unipolar electrode was placed in layer 2/3 (20 mA, 1 ms duration, every 10 s). To record miniature EPSC (mEPSC), TTX (1 μM) was added to normal aCSF and cells were held at − 70 mV.
HEK293T Cell Electrophysiology
Patch clamp recordings in HEK293T cells were made with an Axopatch 200 amplifier (Molecular Devices, USA) and data were sampled at 20 kHz and filtered at 10 kHz (− 3 dB) using pCLAMP8.2 software (Molecular Devices, USA). Series resistances of less than 6 MΩ were admitted and compensated 80% with a 10 μs lag time. Leak current was subtracted online using a P/ − 4 protocol and recordings with leak currents over 100 pA at holding potential were discarded. The datasets for each experimental condition were obtained from at least three independent experiments.
Whole-cell patch clamp recordings in voltage-clamp mode were performed to obtain ionic CaV-mediated calcium currents in transfected HEK293T cells. Recording electrodes with resistances between 2 and 5 MΩ were used and filled with internal solution containing (in mM): 134 CsCl, 10 HEPES, 10 EGTA, 4 MgATP, and 1 EDTA, pH 7.3. All recordings were conducted at room temperature (∼23 °C). To record CaV2.1–2 and CaV1.2–3 currents, the external solution contained (in mM) 140 choline chloride, 10 HEPES, 1 MgCl2.6H2O, and 2 CaCl2.H2O, pH 7.4. To evoke these calcium currents, the test-pulse protocol consisted square pulses from − 100 to − 10 mV (CaV1.3), 0 mV (CaV1.2), or 10 mV (CaV2.1–2) (30 ms duration) followed by a step at − 60 mV (10 ms duration), every 10 s. Current–voltage (I-V) curves were constructed for CaV2.2 currents by measuring the peak of the current in response to 30 ms depolarizing steps from − 80 mV to + 40 mV in 10 mV increments from a holding membrane potential of − 100 mV. To record CaV3.1–3 current, we used an external solution that contained (in mM): 10 BaCl2, 1 MgCl2, 10 HEPES, 140 TEA-chloride and 6 CsCl, pH 7.4, and the test-pulse protocol consisted in square pulses applied from − 100 to − 20 mV (for CaV3.1–2) or − 30 mV (for CaV3.3) (200 ms duration), every 10 s. In all cases, cells were held at − 100 mV as resting potential.
FRET Time Course of Intracellular cAMP
FRET time course of cAMP intracellular levels was measured as previously described [40]. Briefly, transfected HEKT Epac-S H187 cells were seeded in 96-well plates at a density of ~ 100 cells per well. Before each experiment was started, cells were washed with 0.9% NaCl twice, and 100 μl of FluoroBrite DMEM (Thermo-Fisher, USA) was added to each well before placing the plate in a FlexStation 3 (Molecular Devices, USA) at 37 °C. To determine the intracellular cAMP response, the baseline fluorescence signal detected at 475 nm (donor) and 530 nm (FRET) emission with excitation at 430 nm was measured. Using the on-board pipettor, 50 μl of dopamine and/or SCH-23390 3 μM stock solution (to reach a final concentration of 1 μM in the well) or FluoroBrite DMEM was added after 40 s, and then the signals were monitored every 20 s for a total of 600 s. FRET and donor intensities were measured for each time point. FRET/ donor ratio was calculated and normalized to basal levels before stimulation (R/R0) for each time point. An area under the curve (AUC) value of 9-min (540 s) R/R0 intracellular cAMP response was calculated for each replicate. The datasets for each experimental condition were obtained from at least three independent experiments.
Statistical Analysis
Data were analyzed and visualized using OriginPro 8 (Origin-Lab Corp, USA) and Prism 6 (GraphPad Software Inc., USA) software. Figures and artwork were created using Prism 6 (GraphPad Software Inc., USA) and Adobe Illustrator (Adobe Systems Inc., USA). We used the Kolmogorov–Smirnov test for conformity to a normal distribution, and variance homogeneity was examined using Bartlett’s (normally distributed data) and Brown–Forsythe’s (nonnormally distributed data) tests. For normally distributed data, p values were calculated from Student’s one-sample t test or unpaired two-sampled t test and multiple comparison one-way ANOVA with Dunn’s or Tukey’s post hoc tests. In data sets with a nonnormal distribution, p values were calculated from Mann–Whitney test or Kruskal–Wallis test with Dunn’s or Tukey’s post hoc tests. I–V curves were fitted using the linear Boltzman function. The specific statistical test used and sample size for each dataset are indicated in the figure legends, including p values. Data were expressed as the mean ± SEM, and dots represent individual data.
Results
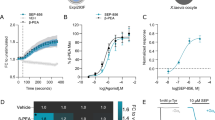
We first assayed the effect of systemically-injected CPZ (1 mg/kg IP) on CaV total native currents of layer 5/6 pyramidal neurons from the mPFC. In order to explore the effect of CPZ on HVA and LVA calcium currents, we measured the current evoked by two different square-pulse voltage protocols, one from − 80 mV to − 30 mV (for LVA currents) and another from − 80 mV to 0 mV (for HVA currents) [41]. We found that CPZ significantly reduces HVA currents and has no effect on LVA currents (Fig. 1a). In order to discriminate the putative inverse agonist effect of CPZ on D1R-like from possible CPZ effects on other cellular targets, we combined the application of CPZ with SCH-23390, a D1R-like selective antagonist that is frequently used in competition binding assays due to its high affinity for these receptors [42]. Specifically, we intra-mPFC administered SCH-23390 (1 μg/mouse), CPZ (1 μg/mouse), or vehicle before systemic injections of CPZ (1 mg/kg, IP) or vehicle (Fig. 1b). We found that IP-injected and intra-mPFC-injected CPZ display undistinguishable effects on native HVA currents, suggesting that this current reduction is due to the local effect of CPZ in the mPFC. Moreover, we observed that SCH-23390 does not affect HVA currents but prevents the effect of IP CPZ on these currents (Fig. 1c). These results could indicate that CPZ prevents D1R-like constitutive stimulation of native HVA currents. To gain further insight on this effect, we examined CPZ and D1R-like constitutive activity’s modulation of the different CaV subtypes expressed in mPFC neurons.

Chlorpromazine selectively reduces high-voltage activated calcium currents in mPFC slices, and this effect is prevented by coadministration of a highly selective D1R-like antagonist. a Representative traces and averaged native calcium currents (ICav) recorded at 0 mV (high voltage-activated (HVA)) or recorded at − 30 mV (low voltage-activated (LVA)) from layer 5/6 pyramidal neurons in mPFC slices obtained from mice IP treated with 0.9% NaCl vehicle (Veh; HVA: n = 11; LVA: n = 14) or chlorpromazine (CPZ, 1 mg/kg; HVA: n = 10; LVA: n = 12). Mann–Whitney test (HVA currents). Unpaired t test (LVA currents). b Schematic representation of the experimental design combining intra-mPFC infusions of aCSF (Veh), SCH-2339 (SCH, 1 µg/brain), or CPZ (1 µg/brain), with IP injections of 0.9% NaCl (Veh) or CPZ (1 mg/kg). c Averaged HVA ICav recorded at 0 mV in layer 5/6 pyramidal neurons in mPFC slices obtained from the experimental conditions shown in B (Veh/Veh, n = 10; SCH/Veh, n = 6; Veh/CPZ, n = 8; CPZ/Veh, n = 13; SCH/CPZ, n = 5). Kruskal–Wallis test with Dunnet’s post test (Veh/Veh). *p < 0.05; **p < 0.005; non-statistically significant (n.s.)
We previously reported that D1R constitutive activity stimulates CaV2.2 currents in a heterologous expression system [14]. Thus, we tested if CaV2.2, one of the HVA channels highly expressed in the mPFC, is a target of CPZ. We assayed the effect of a specific CaV2.2 blocker, ω-conotoxin GVIA (1 µM), in the same experimental conditions shown in Fig. 1b. We found that CPZ, whether it is administered IP or intra-mPFC, dramatically reduces the percentage of current sensitive to ω-conotoxin GVIA in mPFC neurons, and this effect is occluded by the previous intra-mPFC application of SCH-23390 (Fig. 2a). Likewise, we registered CaV2.2 currents in transfected HEK293T cells and applied the same pharmacological strategy preincubating cells with SCH-23390 (1 μM) and/or CPZ (1 μM) (Fig. 2b). We found, as we had previously reported [14], that D1R coexpression increases CaV2.2 currents and that preincubation with the inverse agonist CPZ occludes this upregulation. Interestingly, coincubation with SCH-23390 prevents CPZ inhibitory effect. Additionally, we constructed I–V curves for recombinant CaV2.2 currents in the absence or presence of D1R and found that D1R coexpression drastically reduces the Gmax value and produces a mild but significant shift in the V0.5 value. Moreover, CPZ preincubation occludes D1R effect on Gmax but not on the V0.5 value (Fig. 2c). Furthermore, we verified SCH-23390 antagonism on D1R activity in our experimental system by assaying its effect on dopamine-stimulated cAMP response in cells expressing D1R (Fig. 3a) and dopamine-evoked CaV2.2 current inhibition in cells coexpressing D1R and CaV2.2 (Fig. 3b). These results indicate that CaV2.2 contribute to the native HVA currents inhibited by CPZ in mPFC neurons and that this effect involves a reduction of D1R-like constitutive activity.

D1R-like constitutive activity increases CaV2.2 currents in mPFC neurons and HEK293T cells and this effect is prevented by a highly selective D1R-like antagonist. a Representative traces and averaged values of percentage inhibition of native calcium currents (ICav) recorded at 0 mV (high voltage-activated (HVA)) after bath application of ω-conotoxin GVIA (1 µM) in layer 5/6 pyramidal neurons of mPFC slices obtained from mice cotreated with intra-mPFC infusions of aCSF vehicle (Veh), SCH-2339 (SCH, 1 µg/brain), or chlorpromazine (CPZ, 1 µg/brain) and IP injections of 0.9% NaCl (Veh) or CPZ (1 mg/kg) (Veh/Veh, n = 9; SCH/Veh, n = 4; Veh/CPZ, n = 6; CPZ/Veh, n = 8; SCH/CPZ, n = 4). Kruskal–Wallis test with Dunnet’s post-test (Veh/Veh). b Representative traces and averaged calcium CaV2.2 currents (ICav2.2) recorded in HEK293T cells transfected with CaV2.2 and the auxiliary subunits CaVα2δ1 and CaVβ3, alone (− D1R, n = 24) or with D1R (+ D1R, n = 20) preincubated or not with CPZ (1 µM) and/or SCH-23390 (1 µM) (+ CPZ, n = 12; + SCH, n = 16; + SCH + CPZ, n = 11). Kruskal–Wallis test with Dunnet’s post-test (− D1R). c Averaged calcium current peak-voltage (I-V) relationship (left) and averaged Gmax, V0.5, Vrev, and K values (right) obtained from HEK293T cells cotransfected with CaV2.2 and the auxiliary subunits CaVα2δ1 and CaVβ3, alone (− D1R, n = 21) or with D1R (+ D1R, n = 9) preincubated or not with CPZ (1 µM, n = 5). Mann–Whitney test. *p < 0.05; **p < 0.005; ***p < 0.0005; non-statistically significant (n.s.)

SCH-23390 antagonizes dopamine-evoked cAMP responses and CaV2.2 inhibition. a Left: real-time cAMP time courses recorded in HEK293T cells transfected with D1R or empty plasmid (− D1R) with acute application of dopamine (+ DA, 1 µM), SCH-23390 (+ SCH, 1 µM), and/or DMEM (Veh). Right: averaged values of area under the curve (AUC) measured at 9 min in these experimental conditions (n = 6 for each condition). Kruskal–Wallis test with Dunnet’s post-test (− D1R + Veh). Only significant differences are shown. b Time courses, representative traces, and averaged values of percentage inhibition of ICav2.2 obtained from HEK293T cells cotransfected with CaV2.2, the auxiliary subunits CaVα2δ1 and CaVβ3 and D1R after bath application of dopamine (+ DA, 10 µM) in the absence (+ D1R, n = 4) or presence of SCH-23390 (+ D1R + SCH, 10 µM, n = 4). Mann–Whitney test. *p < 0.05
Other groups of CaV that contribute to HVA currents and are abundantly expressed at soma and dendrites of mPFC neurons are CaV1.2 and CaV1.3 [43, 44]; therefore, we explored if the in vivo administration of CPZ impacts CaV1 currents. We measured the sensitivity of native HVA calcium currents to nifedipine (10 µM), a L-type channel blocker of the dihydropyridine family [45, 46], in layer 5/6 pyramidal neurons of the mPFC. Figure 4a shows that IP or intra-mPFC administration of CPZ significantly reduces the percentage of current sensitive to nifedipine. Surprisingly, the pretreatment with SCH-23390 fails to reverse this effect, suggesting that the effect of CPZ on CaV1 currents involves a D1R-independent mechanism. To test this possibility, we assayed if D1R coexpression impacts CaV1.2 and CaV1.3 currents in transfected HEK293T cells. We found that the presence or the absence of D1R does not affect CaV1.2 or CaV1.3-mediated current levels (Fig. 4b, c). In light of these results and previous work that show an inhibitory effect of CPZ on CaV1 currents in cultured neuroblastoma cells [47], we tested if CPZ has a direct inhibitory effect on CaV1.3 currents. First, we transfected HEK293T cells with the CaV1.3 alone, preincubated them with CPZ (1 µM) for 44 h and found that this chronic CPZ preincubation dramatically reduces basal current levels (Fig. 4d). Also, we evaluated the effect of acute bath application of CPZ (1 µM) on CaV1.3 currents and observed a significant inhibition (Fig. 4e), again in the absence of D1R. Our results suggest that CaV1 channels are not sensitive to D1R constitutive activity and that they are directly inhibited by CPZ.

D1R coexpression has no effect on CaV1 currents while chlorpromazine has a direct inhibitory effect on CaV1 currents. a Representative traces and averaged values of percentage inhibition of native calcium currents (ICav) recorded at 0 mV (high voltage-activated (HVA)) after bath application of nifedipine (10 µM) in layer 5/6 pyramidal neurons of mPFC slices obtained from mice cotreated with intra-mPFC infusions of aCSF vehicle (Veh), SCH-2339 (SCH, 1 µg/brain), or chlorpromazine (CPZ, 1 µg/brain) and IP injections of 0.9% NaCl (Veh) or CPZ (1 mg/kg) (Veh/Veh, n = 9; Veh/CPZ, n = 8; SCH/Veh, n = 3; CPZ/Veh, n = 3; SCH/CPZ, n = 4). Kruskal–Wallis test with Dunnet’s posttest (Veh/Veh). b Representative traces and averaged calcium CaV1.2 currents (ICav1.2) recorded in HEK293T cells transfected with CaV1.2 and the auxiliary subunits CaVα2δ1 and CaVβ3, alone (− D1R, n = 9) or with D1R (+ D1R, n = 6). Mann–Whitney test. c Representative traces and averaged calcium CaV1.3 currents (ICav1.3) recorded in HEK293T cells transfected with CaV1.3 and the auxiliary subunits CaVα2δ1 and CaVβ3, alone (− D1R, n = 23) or with D1R (+ D1R, n = 24). Mann–Whitney test. d Representative traces and averaged ICav1.3 recorded in HEK293T cells transfected with CaV1.3 and the auxiliary subunits CaVα2δ1 and CaVβ3, preincubated or not (− CPZ, n = 13) with chlorpromazine (+ Chronic CPZ, 1 µM, n = 13). Mann–Whitney test. e Representative traces, time course, and averaged values of percentage inhibition of ICav1.3 after bath application of CPZ (+ acute CPZ, 1 µM) in HEK293T cells transfected with CaV1.3 and the auxiliary subunits CaVα2δ1 and CaVβ3 (n = 6). One-sample t test versus zero. *p < 0.05; **p < 0.005; ***p < 0.0001; nonstatistically significant (n.s.)
We explored the contribution of the typically presynaptic channel CaV2.1 to the HVA currents of mPFC neurons. We found a nonsignificant contribution of CaV2.1 to the native HVA current, measured as the percentage of current sensitive to ω-agatoxin-IVA (1 µM) in animals IP treated either with vehicle or CPZ (Fig. 5a), an expected result for our brain slice preparation where most axon terminals of pyramidal neurons are likely absent. Moreover, we confirmed that CaV2.1 is not sensitive to D1R coexpression in HEK293T cells (Fig. 5b). Additionally, we tested the sensitivity of HVA currents from vehicle or CPZ-treated mice to the CaV2.3 blocker SNX-482 (100 nM) and failed to observe a significant effect of this drug on either condition (vehicle: 8.26 ± 4.01%, n = 4, one-sample t test versus zero, p = 0.13; CPZ: 8.28 ± 3.87%, n = 5, one-sample t test versus zero, p = 0.10).

D1R coexpression does not modify CaV2.1 currents in HEK293T cells. a Representative traces and averaged values of percentage inhibition of native calcium currents (ICav) recorded at 0 mV (high voltage-activated (HVA)) after bath application of ω-agatoxin IVA (1 µM; Veh, n = 5; CPZ IP, n = 5) in layer 5/6 pyramidal neurons of mPFC slices obtained from mice treated with IP injections of 0.9% NaCl vehicle (Veh) or chlorpromazine (CPZ, 1 mg/kg). One-sample t test versus zero. b Representative traces and averaged calcium CaV2.1 currents (ICav2.1) recorded in HEK293T cells transfected with CaV2.1, the auxiliary subunits CaVα2δ1 and CaVβ3, alone (− D1R, n = 27) or with D1R (+ D1R, n = 23). Mann–Whitney test. Nonstatistically significant (n.s.)
Finally, we explored the effect of IP administration of CPZ on the main conductances supporting calcium currents at low voltages, which are the CaV3.1, CaV3.2, and CaV3.3 subtypes. We evoked LVA currents with a square-pulse protocol from − 80 mV to − 30 mV, a potential at which we expect a maximum contribution of CaV3 currents. We acutely applied NiCl2 (nickel, 100 µM), which blocks CaV3 and also CaV2.3 currents [38, 39] and found that systemic CPZ treatment does not affect the current sensitivity to nickel (vehicle: 60.57 ± 9.10%, n = 9; CPZ: 61.44 ± 9.25%, n = 8; unpaired t test, p = 0.95). At the low voltages used in these experiments, we expect the majority of the current sensitive to nickel to be produced by CaV3 channels, since CaV2.3 current amplitude at − 30 mV is ~ 20% of its maximum activation [38]. In order to specifically target CaV3 channels, we evaluated the effect of the CaV3 blocker TTA-P2 (1 µM) in neurons from vehicle or CPZ-treated animals and isolated the TTA-P2-sensitive LVA currents by subtracting current remaining in the presence of TTA-P2 from control current. We observed a similar sensitivity of LVA currents in both experimental conditions, and this TTA-P2-sensitive current displayed a faster kinetic, consistent with a CaV3-mediated current (Fig. 6a). Then, we assayed the effect of D1R coexpression on CaV3.1, CaV3.2 and CaV3.3 channels in transfected HEK293T cells, and found that neither of these currents is modified by D1R coexpression (Fig. 6b–d).

D1R coexpression does not modify CaV3 currents in mPFC neurons or HEK293T cells. a Representative traces and averaged values of percentage inhibition of native calcium currents (ICav) recorded at − 30 mV (low voltage-activated (LVA)) after bath application of TTA-P2 (1 µM) in layer 5/6 pyramidal neurons of mPFC slices obtained from mice treated with IP injections of 0.9% NaCl vehicle (Veh, n = 7) or chlorpromazine (CPZ, 1 mg/kg, n = 5). Unpaired t test. b Representative traces and averaged calcium CaV3.1 currents (ICav3.1) recorded in HEK293T cells transfected with CaV3.1 alone (− D1R, n = 13) or with D1R (+ D1R, n = 15). Mann–Whitney test. c Representative traces and averaged calcium CaV3.2 currents (ICav3.2) recorded in HEK293T cells transfected with CaV3.2 alone (− D1R, n = 11) or with D1R (+ D1R, n = 13). Unpaired t test. d Representative traces and averaged calcium CaV3.3 currents (ICav3.3) recorded in HEK293T cells transfected with CaV3.3 alone (− D1R, n = 9) or with D1R (+ D1R, n = 10). Unpaired t test. Nonstatistically significant (n.s.)
Since we found that CPZ reduces CaV1 and CaV2.2 currents and these effects could negatively impact somatodendritic calcium currents, we hypothesized that CPZ could alter calcium-dependent neuronal functions, such as the excitatory postsynaptic responses [48]. Since the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor density on postsynaptic sites is controlled by calcium dependent mechanisms, we evaluated if CPZ treatment modifies the spontaneous excitatory synaptic activity and the excitatory response of mPFC neurons to electrical stimuli. We explored the effect of CPZ IP treatment on mEPSCs and compared their amplitude, a parameter generally related to postsynaptic excitatory receptor density, and their frequency, typically associated to the degree of glutamate release from presynaptic vesicles [49]. We found that IP CPZ reduces both parameters suggesting the presence of a postsynaptic CPZ effect, possibly due the reduction of somatodendritic CaV1 and CaV2.2 currents, as well as an additional mechanism of CPZ at the presynaptic level (Fig. 7a). Next, we studied the effect of IP CPZ on the evoked EPSC recorded in layer 5/6 and evoked by electrical stimulation in layer 2/3 and found a significant reduction of the EPSC amplitude (Fig. 7b). Meanwhile, since it has been reported that acute D1R-like agonist application potentiates EPSCs in the mPFC [50], we evaluated the effect of SKF-38393 on EPSC. We confirmed that this D1R-like agonist increases the EPSC amplitude in our experimental setup, although we failed to observe a significant impact of CPZ pretreatment on this potentiation (vehicle: 41.49 ± 20.99% of increase, n = 3; vs. CPZ: 29.37 ± 8.75% of increase, n = 4; Mann–Whitney test, p > 0.999). Another possible consequence of a reduced activity of CaV1 and CaV2.2 could be a smaller response of transcriptional activity upon stimulation. Considering that SKF-38393 increases the levels of the transcriptional factor c-Fos [51], we evaluated if this effect persists with a CPZ pretreatment. Therefore, we performed immunohistochemistry assays to evaluate the number of c-Fos-IR cells in the mPFC after IP treatment with vehicle, CPZ, and/or SKF-38393 (Fig. 8a). In a set of mice, we confirmed that SKF-38393 treatment (3 mg/kg) increases the number of c-Fos-IR cells in the mPFC, as compared to vehicle treatment (vehicle: 52.31 ± 9.69 c-Fos IR cells, n = 7; vs. SKF-38393: 134.7 ± 22.30 c-Fos IR cells, n = 9; unpaired t test, p = 0.005) (Fig. 8b). Next, we compared c-Fos induction in mice pretreated with vehicle or CPZ (1 mg/kg) and, 24 h later, with SKF-38393 (3 mg/kg). We observed that, in the latter case, SKF-38393 treatment does not induce a significant increase in the number of c-Fos-IR cells in the mPFC, as compared to the CPZ/vehicle treatment (CPZ/vehicle: 74.57 ± 10.17 c-Fos IR cells, n = 6; vs. CPZ/SKF-38393: 118.40 ± 16.29 c-Fos IR cells, n = 6; Mann–Whitney test, p = 0.094) (Fig. 8c). Overall, these findings suggest that CPZ administration impacts calcium-dependent processes in the mPFC.

Chlorpromazine decreases basal and evoked excitatory currents in mPFC neurons. a Top: representative traces of miniature excitatory postsynaptic currents (mEPSC) recorded from layer 5/6 pyramidal neurons in mPFC slices obtained from mice IP treated with 0.9% NaCl vehicle (Veh, n = 17) or chlorpromazine (CPZ, 1 mg/kg, n = 14). Bottom: averaged cumulative probability values and mean amplitude or frequency values of mEPSC. Mann–Whitney test. b Left: schematic representation of a coronal brain slice containing the mPFC indicating the position of the stimulating electrode (SE) in layer 2/3 and the recording electrode (RE) in layer 5/6. Inset: infrared differential interference contrast image of a pyramidal neuron from layer 5/6 of the mPFC under experimental conditions. Scale bar represents 10 µm. Middle and right: representative traces and averaged amplitude of excitatory postsynaptic currents (EPSC) evoked by electrical stimuli and recorded from layer 5/6 pyramidal neurons in mPFC slices obtained from mice IP treated with 0.9% NaCl (Veh, n = 9) or chlorpromazine (CPZ, 1 mg/kg, n = 12). Black triangles indicate the electrical stimulus application. Mann–Whitney test

SKF-38393 enhances c-Fos reactivity in the mPFC, and this effect is impaired by chlorpromazine pretreatment. a Schematic representation of a coronal brain slice containing the mPFC. Inset shows the mPFC area delimitated according to Paxinos and Franklin [34]. Fmi: forceps minor corpus callosum. b Representative microphotographs of the immunohistochemistry against c-Fos in coronal brain slices containing the mPFC of mice IP treated with 0.9% NaCl vehicle (Veh) or SKF-38393 (SKF, 3 mg/kg) 2 h before sacrifice. c Representative microphotographs of the immunohistochemistry against c-Fos in coronal brain slices containing the mPFC of mice IP treated with chlorpromazine (CPZ, 1 mg/kg) 24 h before IP injections of 0.9% NaCl vehicle (Veh) or SKF-38393 (SKF, 3 mg/kg) (2 h before sacrifice). Schematic divisions according to Paxinos and Franklin [34] are overlayed in the figures. Scale bar: 100 µm
Discussion
In this study, we found that CPZ, a commonly used antipsychotic drug and a D1R-like inverse agonist, reduces HVA currents of pyramidal neurons from the mouse mPFC, with no effect on LVA currents. We found that CPZ reduced D1R-like constitutive activity-mediated stimulation of CaV2.2 currents in HEK293T cells and mPFC neurons. Additionally, CPZ directly inhibited CaV1 currents. Both mechanisms of action contribute to the reduced HVA currents. In contrast, neither CPZ significantly affected neuronal CaV2.1, CaV2.3, or CaV3 conductances, nor D1R constitutive activity modified the recombinant CaV2.1 or CaV3 currents. We evaluated the effect of CPZ on some neuronal functions that could be altered by a lessened postsynaptic CaV1 and CaV2.2 activity. We found that this antipsychotic drug significantly reduced both spontaneous and evoked excitatory postsynaptic currents and impaired the transcriptional activity induced by a specific D1R-like agonist in mPFC neurons.
Optimal levels of D1R-like stimulation are key for prefrontal-mediated cognition and D1R-based therapeutics have been proposed for the treatment of cognitive deficits caused by schizophrenia and other psychiatric disorders [52]. Difficulties have arisen in determining what the optimal level of D1R-stimulation is and, in this regard, the D1R-like constitutive activity could be a factor that is not being taken into account. Abundant evidence shows that physical and cognitive exercise can modulate dopamine levels and D1R-like expression in the PFC and prove beneficial on cognition [53,54,55]. This dynamic levels of D1R-like expression in the PFC could mean different levels of constitutive signaling. Additionally, understanding the physiological relevance of constitutive activity would require to individually study the cellular targets of this signaling, without presuming that the known targets of D1R-like dopamine-evoked activity will be equally sensitive to its constitutive activity. In this work, we focused on CaV subtype sensitivity to D1R-like constitutive activity, knowing that these proteins are sensitive to other constitutively active GPCRs and that CaV activity is key for many neuronal functions.
D1R-like agonist-evoked activity differentially modulates CaV subtypes. The rapid activation of D1R-like induced by dopamine or subtype-specific agonists, such as SKF-38393 used in this study, acutely inhibits CaV2.1, CaV2.2, and CaV3 channels [33, 56,57,58], whereas it enhances CaV1 currents [57, 58]. However, less is known about how D1R-like constitutive activity affects CaV currents. Here, we used a pharmacological strategy combining a D1R-like inverse agonist (CPZ) and neutral antagonist (SCH-23390) to manipulate the receptor constitutive activity and its effect on CaV currents. Regarding the mechanism of action of these ligands, it is claimed that inverse agonists stabilize the inactive state and reduce basal GDP/GTP exchange, while agonists bind and stabilize a specific active state of a given GPCR. In contrast, neutral antagonists do not alter the equilibrium between active and inactive states nor do they change basal G-protein activity, but instead they block the inhibitory effect of inverse agonists and the stimulatory effect of agonists [19]. We confirmed that SCH-23390 effectively antagonized dopamine-induced D1R activation in our experimental setup using electrophysiological recordings and cAMP measurements in a heterologous expression system, similar to what was previously shown [25]. Here, we found that SCH-23390 successfully prevented CPZ effect on D1R constitutive activity, administered both in vivo and in vitro. Thanks to SCH-23390 high specificity and binding affinity to D1R-like [42, 59], this pharmacological approach allowed us to elucidate whether CPZ effects were acting on D1R-like constitutive activity or on other cellular targets. The same strategy was previously used in a study that measured adenylyl cyclase activity in PC2 cells transfected with D1R and treated with the inverse agonists clozapine and fluphenazine [25]. More recently, SCH-23390 was shown to occlude the inverse agonism of flupentixol and clozapine on D5R constitutive activity in striatal slices from a mouse model of dyskinesia [16]. We confirmed that CPZ acts locally in the mPFC reducing D1R-like constitutive upregulation of CaV2.2 currents. Interestingly, CPZ effect on native CaV1 currents was insensitive to SCH-23390 administration in the mPFC. In addition, our data in transfected HEK293T cells show that D1R coexpression does not modify CaV1.2–3 currents and that CPZ acutely and chronically reduces CaV1.3, indicating that this drug has a direct inhibitory effect on these channels. Likewise, a previous work showed that CPZ had a direct and reversible blocking action on calcium currents from cultured mouse neuroblastoma cells [47].
We found that CPZ reduces HVA currents in mPFC neurons through two independent mechanisms. First, CPZ affects CaV2.2 contribution through a D1R-dependent mechanism: while D1R constitutive activity enhances CaV2.2 currents through an increase in the Gmax value, CPZ acting as a D1R-like inverse agonist occludes this effect and reduces these currents. Interestingly, CPZ preincubation was able to prevent D1R increase of the Gmax value but not the observed shift in the V0.5 value, suggesting that this effect of D1R could be due to a different mechanism. Secondly, CPZ significantly reduces CaV1 currents in the mPFC through a direct effect on these channels that is independent of D1R. Since CaV1 are typically postsynaptic channels [44] and CaV2.2 have been reported to have a noncanonical postsynaptic distribution in the mPFC [33], both of these channels would control somatodendritic calcium entry in pyramidal mPFC neurons. In contrast, CPZ did not affect CaV2.1, CaV2.3, and CaV3, which are expected to contribute to neurotransmitter release and firing, respectively [60,61,62,63]. Thus, we postulate that D1R-like constitutive activity could have an impact on calcium-dependent postsynaptic activity. Considering that CPZ effect is partially due to its D1R-like inverse agonist properties, our results suggest that D1R-like constitutive activity is related to higher basal excitatory responses, larger EPSC responses to electrical stimuli, and increased transcriptional activity induced by a D1R-like agonist. These larger excitatory responses would produce bigger somatic and dendritic depolarizations that could trigger postsynaptic plasticity mechanisms including increased AMPA membrane expression and phosphorylation, among other long-lasting changes at the transcriptional and structural level [64, 65]. On the other hand, the mPFC is modulated by dopamine release from ventral tegmental area projections [66]. Previous reports demonstrated that the activation of D1R-like by dopamine or SKF-38393 increases EPSC in the PFC, the entorhinal cortex, and the striatum [50, 67, 68]. Here, we found that SKF-38393 application potentiated the EPSCs in neurons from control and CPZ-treated mice. Moreover, several studies showed that D1R-like agonists stimulate c-Fos reactivity in the mPFC [69,70,71] and other brain areas expressing D1R, such as the hypothalamus [51, 72]. In this work, we demonstrated that CPZ pretreatment impairs the SKF-38393-induced transcriptional activity. Taken together, these results allow us to propose that mPFC neuron responses to glutamatergic and dopaminergic inputs are altered by CPZ treatment and could therefore depend on D1R-like constitutive activity levels. More experiments are required to determine the involvement of D1R constitutive activity on these observations.
Isolating the effect of CPZ as an inverse agonist of D1R-like activity in brain slices is challenging. CPZ is a fairly promiscuous drug, and it was reported to act on cellular targets other than D1R-like and D2R-like, such as serotonin receptors 2A (5-HT2A), adrenergic receptors α1A and α1B, muscarinic M2 autoreceptors, and GABA receptors [73,74,75,76]. Thus, in the mPFC, there could be compensations of D1R and CPZ effects and cross-talk between their signaling pathways. In this regard, combining the native system of mPFC slices with the heterologous expression system of HEK293T cells provided us with a tool to isolate and thoroughly study this effect in a highly controlled system. In this work, we were able to reproduce in HEK293T cells the results seen in mPFC slices by cotransfecting cells only with the selected cDNA, suggesting that other receptors are not required for the observed effects. Moreover, CPZ is a potent first-generation antipsychotic that maintains its clinical relevance after 70 years from its discovery [26]. Even though CPZ has many active metabolites that are able to cross the blood–brain barrier, it was shown that unaltered CPZ is the main responsible for the effects at the central nervous system [77]. The high costs and times required for new medications to reach the bedside make repurposing or repositioning of old drugs an appealing strategy [78]. For example, CPZ showed a neuroprotective role, ameliorating the blood–brain barrier disruption, inflammation, and apoptosis in a rat model of ischemic stroke [79]. Recent studies identified CPZ as a candidate for repurposing for acute myeloid leukemia therapy, showing the antileukemic activities of CPZ administration in vitro and in vivo in mice and zebrafish larvae [80, 81]. Additionally, CPZ was proposed as a possible treatment against breast cancer, malignant gliomas, and helminthic parasite infection [78, 82, 83]. Our results describing a noncanonical action of CPZ as an inverse agonist of D1R-like may contribute to further understanding the action mechanisms of CPZ therapeutic and adverse effects, as well as for the repurposing of this already available drug.
A great research effort is being dedicated to the discovery and development of better or new D1R ligands with improved pharmacokinetics and selectivity that could be tested in the treatment of psychiatric disorders like schizophrenia [53, 84].Various D1R-like agonists have been reported, and some were even tested in patients showing promising results, but they have failed development due to low bioavailability, tolerance, and D2R side effects, among other reasons [52, 53, 85]. For example, dihydrexidine was the first centrally available full D1R-like agonist, and it has been tested in patients with schizophrenia and Parkinson disease, but it showed severe limitations like a low oral bioavailability and rapid metabolization [86, 87]. Other approaches of modulating D1R activity have been studied as possible treatments for psychiatric disease, including the pursuit of partial agonists, antagonists, and positive allosteric modulators [85, 88]. For instance, ecopipam (or SCH-39166), an analog to the SCH-23390 used in this work, is a D1R-like antagonist that was tested in clinical trials for the treatment of schizophrenia, drug abuse, and obesity [89,90,91]. In this regard, the development, identification, and study of D1R-like inverse agonists could be significant for the treatment of psychiatric diseases.
Data Availability
Original data are available from the corresponding author on reasonable request.
References
Cools R (2008) Role of dopamine in the motivational and cognitive control of behavior. Neuroscientist 14(4):381–395. https://doi.org/10.1177/1073858408317009
Leal E et al (2013) Effects of dopaminergic system activation on feeding behavior and growth performance of the sea bass (Dicentrarchus labrax): a self-feeding approach. Horm Behav 64(1):113–121. https://doi.org/10.1016/j.yhbeh.2013.05.008
Beaulieu JM, Espinoza S, Gainetdinov RR (2015) Dopamine receptors–IUPHAR review 13. Br J Pharmacol 172(1):1–23. https://doi.org/10.1111/bph.12906
Beaulieu J et al (2019) Dopamine receptors (version 2019.4) in the IUPHAR/BPS guide to pharmacology database. IUPHAR/BPS Guide Pharmacol. https://doi.org/10.2218/gtopdb/F20/2019.4
Abi-Dargham A et al (2002) Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci 22(9):3708–3719. https://doi.org/10.1523/JNEUROSCI.22-09-03708.2002
Tiberi M, Caron MG (1994) High agonist-independent activity is a distinguishing feature of the dopamine D1B receptor subtype. J Biol Chem 269(45):27925–27931
Abraham AD, Neve KA, Lattal KM (2016) Activation of D1/5 dopamine receptors: a common mechanism for enhancing extinction of fear and reward-seeking behaviors. Neuropsychopharmacology 41(8):2072. https://doi.org/10.1038/npp.2016.5
Centonze D et al (2003) Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J Neurosci 23(24):8506–8512. https://doi.org/10.1523/JNEUROSCI.23-24-08506.2003
Vijayraghavan S et al (2007) Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci 10(3):376. https://doi.org/10.1038/nn1846
Zhang B et al. (2014) Constitutive activities and inverse agonism in dopamine receptors, in Advances in pharmacology. Elsevier. pp 175–214. https://doi.org/10.1016/B978-0-12-417197-8.00007-9
Zhang B, Yang X, Tiberi M (2015) Functional importance of two conserved residues in intracellular loop 1 and transmembrane region 2 of Family A GPCRs: insights from ligand binding and signal transduction responses of D1 and D5 dopaminergic receptor mutants. Cell Signal 27(10):2014–2025. https://doi.org/10.1016/j.cellsig.2015.07.006
Plouffe B et al. (2010) Probing the constitutive activity among dopamine D1 and D5 receptors and their mutants, in Methods in enzymology, Elsevier. pp 295–328. https://doi.org/10.1016/B978-0-12-381298-8.00016-2
Chetrit J et al (2013) Inhibiting subthalamic D5 receptor constitutive activity alleviates abnormal electrical activity and reverses motor impairment in a rat model of Parkinson’s disease. J Neurosci 33(37):14840–14849. https://doi.org/10.1523/JNEUROSCI.0453-13.2013
McCarthy CI, et al (2020) Constitutive activity of dopamine receptor type 1 (D1R) increases CaV2. 2 currents in PFC neurons. J Gen Physiol 152(5). https://doi.org/10.1085/jgp.201912492
Wang Q et al (2020) Constitutive activity of a G protein-coupled receptor, DRD1, contributes to human cerebral organoid formation. Stem Cells 38(5):653–665. https://doi.org/10.1002/stem.3156
Paz RMS, Agostina M, Rela L, Murer MG, Tubert C (2022) D1/D5 inverse agonists restore striatal cholinergic interneuron physiology in dyskinetic mice. Mov Disord. https://doi.org/10.1002/mds.29055
Koski G, Streaty R, Klee W (1982) Modulation of sodium-sensitive GTPase by partial opiate agonists. An explanation for the dual requirement for Na+ and GTP in inhibitory regulation of adenylate cyclase. J Biol Chem 257(23):14035–14040
Beinborn M et al (2004) Ligand function at constitutively active receptor mutants is affected by two distinct yet interacting mechanisms. Mol Pharmacol 65(3):753–760. https://doi.org/10.1124/mol.65.3.753
Seifert R, Wenzel-Seifert K (2002) Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol 366(5):381–416. https://doi.org/10.1007/s00210-002-0588-0
Lopez Soto EJ et al (2015) Constitutive and ghrelin-dependent GHSR1a activation impairs CaV2.1 and CaV2.2 currents in hypothalamic neurons. J Gen Physiol 146(3):205–219. https://doi.org/10.1085/jgp.201511383
Rojo D et al (2020) Mouse models for V103I and I251L gain of function variants of the human MC4R display decreased adiposity but are not protected against a hypercaloric diet. Mol Metab. 42:101077. https://doi.org/10.1016/j.molmet.2020.101077
Torz LJ et al (2020) Metabolic insights from a GHSR-A203E mutant mouse model. Mol Metab 39:101004. https://doi.org/10.1016/j.molmet.2020.101004
Nijenhuis WA, Oosterom J, Adan RA (2001) AgRP (83–132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol Endocrinol 15(1):164–171. https://doi.org/10.1210/mend.15.1.0578
Mkadmi C et al (2018) N-terminal liver-expressed antimicrobial peptide 2 (LEAP2) region exhibits inverse agonist activity toward the ghrelin receptor. J Med Chem 62(2):965–973. https://doi.org/10.1021/acs.jmedchem.8b01644
Cai G et al (1999) Inverse agonist properties of dopaminergic antagonists at the D1A dopamine receptor: uncoupling of the D1Adopamine receptor from Gs protein. Mol Pharmacol 56(5):989–996. https://doi.org/10.1124/mol.56.5.989
Boyd-Kimball D et al (2018) Classics in chemical neuroscience: chlorpromazine. ACS Chem Neurosci 10(1):79–88. https://doi.org/10.1021/acschemneuro.8b00258
Dolphin AC (2016) Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol 594(19):5369–5390. https://doi.org/10.1113/JP272262
Woodside B et al (2004) NMDA receptors and voltage-dependent calcium channels mediate different aspects of acquisition and retention of a spatial memory task. Neurobiol Learn Mem 81(2):105–114. https://doi.org/10.1016/j.nlm.2003.10.003
Catterall WA (2011) Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3(8):a003947. https://doi.org/10.1101/cshperspect.a003947
Agosti F et al (2017) Melanocortin 4 receptor constitutive activity inhibits L-type voltage-gated calcium channels in neurons. Neuroscience 346:102–112. https://doi.org/10.1016/j.neuroscience.2017.01.007
Mustafá ER, Gonzalez SC, Raingo J (2019) Ghrelin selectively inhibits Ca V 3.3 subtype of low-voltage-gated calcium channels. Mol Neurobiol:1–14. https://doi.org/10.1007/s12035-019-01738-y.
Cordisco Gonzalez S et al (2020) Dopamine receptor type 2 and ghrelin receptor coexpression alters CaV2.2 modulation by G protein signaling cascades. ACS Chem Neurosci. 11(1):3–13. https://doi.org/10.1021/acschemneuro.9b00426
Kisilevsky AE et al (2008) D1 receptors physically interact with N-type calcium channels to regulate channel distribution and dendritic calcium entry. Neuron 58(4):557–570. https://doi.org/10.1016/j.neuron.2008.03.002
Paxinos G, Franklin KB (2019) Paxinos and Franklin’s the mouse brain in stereotaxic coordinates. Academic press
Cornejo MP et al (2021) Ghrelin treatment induces rapid and delayed increments of food intake: a heuristic model to explain ghrelin’s orexigenic effects. Cell Mol Life Sci 78(19):6689–6708. https://doi.org/10.1007/s00018-021-03937-0
Klarenbeek JB et al (2011) A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PloS one 6(4):e19170. https://doi.org/10.1371/journal.pone.0019170
Wang Y et al (2006) Heterogeneity in the pyramidal network of the medial prefrontal cortex. Nat Neurosci 9(4):534. https://doi.org/10.1038/nn1670
Zamponi G, Bourinet E, Snutch T (1996) Nickel block of a family of neuronal calcium channels: subtype-and subunit-dependent action at multiple sites. J Membr Biol 151(1):77–90. https://doi.org/10.1007/s002329900059
Mlinar B, Enyeart J (1993) Block of current through T-type calcium channels by trivalent metal cations and nickel in neural rat and human cells. J Physiol 469(1):639–652. https://doi.org/10.1113/jphysiol.1993.sp019835
Carozzo A et al (2019) Identification of MRP4/ABCC4 as a target for reducing the proliferation of pancreatic ductal adenocarcinoma cells by modulating the cAMP efflux. Mol Pharmacol 96(1):13–25. https://doi.org/10.1124/mol.118.115444
Müller T, Misgeld U, Swandulla D (1992) Ionic currents in cultured rat hypothalamic neurones. J Physiol 450(1):341–362. https://doi.org/10.1113/jphysiol.1992.sp019130
Neiens P, Höfner G, Wanner KT (2015) MS binding assays for D1 and D5 dopamine receptors. ChemMedChem 10(11):1924–1931. https://doi.org/10.1002/cmdc.201500355
Sinnegger-Brauns MJ et al (2009) Expression and 1, 4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol 75(2):407–414. https://doi.org/10.1124/mol.108.049981
Heng L-J et al (2011) Concurrent upregulation of postsynaptic L-type Ca2+ channel function and protein kinase A signaling is required for the periadolescent facilitation of Ca2+ plateau potentials and dopamine D1 receptor modulation in the prefrontal cortex. Neuropharmacology 60(6):953–962. https://doi.org/10.1016/j.neuropharm.2011.01.041
Lee T-S et al (2006) Actions of mibefradil, efonidipine and nifedipine block of recombinant T-and L-type Ca2+ channels with distinct inhibitory mechanisms. Pharmacology 78(1):11–20. https://doi.org/10.1159/000094900
Striessnig J, Ortner NJ, Pinggera A (2015) Pharmacology of L-type calcium channels: novel drugs for old targets? Curr Mol Pharmacol 8(2):110–122. https://doi.org/10.2174/1874467208666150507105845
Ogata N, Yoshii M, Narahashi T (1990) Differential block of sodium and calcium channels by chlorpromazine in mouse neuroblastoma cells. J Physiol 420(1):165–183. https://doi.org/10.1113/jphysiol.1990.sp017906
Huganir RL, Nicoll RA (2013) AMPARs and synaptic plasticity: the last 25 years. Neuron 80(3):704–717. https://doi.org/10.1016/j.neuron.2013.10.025
Kavalali ET (2015) The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16(1):5–16. https://doi.org/10.1038/nrn3875
Gonzalez-Islas C, Hablitz JJ (2003) Dopamine enhances EPSCs in layer II–III pyramidal neurons in rat prefrontal cortex. J Neurosci 23(3):867–875. https://doi.org/10.1523/JNEUROSCI.23-03-00867.2003
Chocyk A, Czyrak A, Wedzony K (2008) Dopamine D1-like receptors agonist SKF 38393 increases cFOS expression in the paraventricular nucleus of the hypothalamus–impact of acute and chronic cocaine. J Physiol Pharmacol: Off J Polish Physiol Soc 59(3):425–440
Abi-Dargham A et al (2022) Dopamine D1R receptor stimulation as a mechanistic pro-cognitive target for schizophrenia. Schizophr Bull 48(1):199–210. https://doi.org/10.1093/schbul/sbab095
Arnsten AF et al (2017) Novel dopamine therapeutics for cognitive deficits in schizophrenia. Biol Psychiat 81(1):67–77. https://doi.org/10.1016/j.biopsych.2015.12.028
Braun U et al (2021) Brain network dynamics during working memory are modulated by dopamine and diminished in schizophrenia. Nat Commun 12(1):1–11. https://doi.org/10.1038/s41467-021-23694-9
Kintz N, Petzinger GM, Jakowec MW (2017) Treadmill exercise modifies dopamine receptor expression in the prefrontal cortex of the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-lesioned mouse model of Parkinson’s disease. NeuroReport 28(15):987–995. https://doi.org/10.1097/WNR.0000000000000865
Momiyama T, Fukazawa Y (2007) D1-like dopamine receptors selectively block P/Q-type calcium channels to reduce glutamate release onto cholinergic basal forebrain neurones of immature rats. J Physiol 580(1):103–117. https://doi.org/10.1113/jphysiol.2006.125724
Pfeiffer-Linn C, Lasater EM (1993) Dopamine modulates in a differential fashion T-and L-type calcium currents in bass retinal horizontal cells. J Gen Physiol 102(2):277–294. https://doi.org/10.1085/jgp.102.2.277
Surmeier DJ et al (1995) Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14(2):385–397. https://doi.org/10.1016/0896-6273(95)90294-5
Bourne JA (2001) SCH 23390: the first selective dopamine D1-like receptor antagonist. CNS Drug Rev 7(4):399–414. https://doi.org/10.1111/j.1527-3458.2001.tb00207.x
Kisilevsky AE, Zamponi GW (2008) Presynaptic calcium channels: structure, regulators, and blockers. Handb Exp Pharmacol 184:45–75. https://doi.org/10.1007/978-3-540-74805-2_3
Bender KJ et al (2012) Control of firing patterns through modulation of axon initial segment T-type calcium channels. J Physiol 590(1):109–118. https://doi.org/10.1113/jphysiol.2011.218768
Zaitsev A et al (2007) P/Q-type, but not N-type, calcium channels mediate GABA release from fast-spiking interneurons to pyramidal cells in rat prefrontal cortex. J Neurophysiol 97(5):3567–3573. https://doi.org/10.1152/jn.01293.2006
Perez-Reyes E (2003) Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev 83(1):117. https://doi.org/10.1152/physrev.00018.2002
Kennedy MB (2000) Signal-processing machines at the postsynaptic density. Science 290(5492):750–754. https://doi.org/10.1126/science.290.5492.750
Baltaci SB, Mogulkoc R, Baltaci AK (2019) Molecular mechanisms of early and late LTP. Neurochem Res 44(2):281–296. https://doi.org/10.1007/s11064-018-2695-4
Anastasiades PG, Carter AG (2021) Circuit organization of the rodent medial prefrontal cortex. Trends Neurosci 44(7):550–563. https://doi.org/10.1016/j.tins.2021.03.006
Glovaci I, Caruana D, Chapman C (2014) Dopaminergic enhancement of excitatory synaptic transmission in layer II entorhinal neurons is dependent on D1-like receptor-mediated signaling. Neuroscience 258:74–83. https://doi.org/10.1016/j.neuroscience.2013.10.076
Umemiya M, Raymond LA (1997) Dopaminergic modulation of excitatory postsynaptic currents in rat neostriatal neurons. J Neurophysiol 78(3):1248–1255. https://doi.org/10.1152/jn.1997.78.3.1248
Wass C et al (2013) Dopamine D1 sensitivity in the prefrontal cortex predicts general cognitive abilities and is modulated by working memory training. Learn Mem 20(11):617–627. https://doi.org/10.1101/lm.031971.113
Numa C et al (2019) Social defeat stress-specific increase in c-Fos expression in the extended amygdala in mice: involvement of dopamine D1 receptor in the medial prefrontal cortex. Sci Rep 9(1):1–9. https://doi.org/10.1038/s41598-019-52997-7
Felippe RM, et al (2021) Experimental social stress: dopaminergic receptors, oxidative stress, and c-Fos protein are involved in highly aggressive behavior. Front Cell Neurosci:324. https://doi.org/10.3389/fncel.2021.696834.
Bender M, Drago J, Rivkees SA (1997) D1 receptors mediate dopamine action in the fetal suprachiasmatic nuclei: studies of mice with targeted deletion of the D1 dopamine receptor gene. Mol Brain Res 49(1–2):271–277. https://doi.org/10.1016/s0169-328x(97)00161-7
Farnbach-Pralong D et al (1998) Clozapine and olanzapine treatment decreases rat cortical and limbic GABAA receptors. Eur J Pharmacol 349(2–3):R7–R8. https://doi.org/10.1016/s0014-2999(98)00285-4
Gokhale S, Gulati O, Parikh H (1964) An investigation of the adrenergic blocking action of chlorpromazine. Br J Pharmacol Chemother 23(3):508–520. https://doi.org/10.1111/j.1476-5381.1964.tb01606.x
Trichard C et al (1998) Binding of antipsychotic drugs to cortical 5-HT2A receptors: a PET study of chlorpromazine, clozapine, and amisulpride in schizophrenic patients. Am J Psychiatry 155(4):505–508. https://doi.org/10.1176/ajp.155.4.505
Johnson DE et al (2005) The role of muscarinic receptor antagonism in antipsychotic-induced hippocampal acetylcholine release. Eur J Pharmacol 506(3):209–219. https://doi.org/10.1016/j.ejphar.2004.11.015
Alfredsson G, Wiesel F-A, Skett P (1977) Levels of chlorpromazine and its active metabolites in rat brain and the relationship to central monoamine metabolism and prolactin secretion. Psychopharmacology 53(1):13–18. https://doi.org/10.1007/BF00426688
Abbruzzese C et al (2020) Repurposing chlorpromazine in the treatment of glioblastoma multiforme: analysis of literature and forthcoming steps. J Exp Clin Cancer Res 39(1):1–3. https://doi.org/10.1186/s13046-020-1534-z
Guo S et al (2021) An inhibitory and beneficial effect of chlorpromazine and promethazine (C+ P) on hyperglycolysis through HIF-1α regulation in ischemic stroke. Brain Res 1763:147463. https://doi.org/10.1016/j.brainres.2021.147463
Gundersen ET et al (2022) Repurposing chlorpromazine for anti-leukaemic therapy by nanoparticle encapsulation. Int J Pharm 612:121296. https://doi.org/10.1016/j.ijpharm.2021.121296
Rai S et al (2020) Chlorpromazine eliminates acute myeloid leukemia cells by perturbing subcellular localization of FLT3-ITD and KIT-D816V. Nat Commun 11(1):1–14. https://doi.org/10.1038/s41467-020-17666-8
Yang C-E et al (2019) The antipsychotic chlorpromazine suppresses YAP signaling, stemness properties, and drug resistance in breast cancer cells. Chem Biol Interact 302:28–35. https://doi.org/10.1016/j.cbi.2019.01.033
Weeks JC et al (2018) Sertraline, paroxetine, and chlorpromazine are rapidly acting anthelmintic drugs capable of clinical repurposing. Sci Rep 8(1):1–17. https://doi.org/10.1038/s41598-017-18457-w
Teng X et al (2022) Ligand recognition and biased agonism of the D1 dopamine receptor. Nat Commun 13(1):1–11. https://doi.org/10.1038/s41467-022-30929-w
Jones-Tabah J, et al (2021) The signaling and pharmacology of the dopamine D1 receptor. Front Cell Neurosci 15. https://doi.org/10.3389/fncel.2021.806618.
Mu Q et al (2007) A single 20 mg dose of the full D1 dopamine agonist dihydrexidine (DAR-0100) increases prefrontal perfusion in schizophrenia. Schizophr Res 94(1–3):332–341. https://doi.org/10.1016/j.schres.2007.03.033
Blanchet PJ et al (1998) Effects of the full dopamine D1 receptor agonist dihydrexidine in Parkinson’s disease. Clin Neuropharmacol 21(6):339–343
Hall A, Provins L, Valade A (2018) Novel strategies to activate the dopamine D1 receptor: recent advances in orthosteric agonism and positive allosteric modulation. J Med Chem 62(1):128–140. https://doi.org/10.1021/acs.jmedchem.8b01767
Haney M et al (2001) Effects of ecopipam, a selective dopamine D1 antagonist, on smoked cocaine self-administration by humans. Psychopharmacology 155(4):330–337. https://doi.org/10.1007/s002130100725
Astrup A et al (2007) Randomized controlled trials of the D1/D5 antagonist ecopipam for weight loss in obese subjects. Obesity 15(7):1717–1731. https://doi.org/10.1038/oby.2007.205
Khasnavis T et al (2016) A double-blind, placebo-controlled, crossover trial of the selective dopamine D1 receptor antagonist ecopipam in patients with Lesch-Nyhan disease. Mol Genet Metab 118(3):160–166. https://doi.org/10.1016/j.ymgme.2016.04.012
Acknowledgements
We would like to thank Dr. Diane Lipscombe (Brown University, Providence, USA) for providing the CaV1, CaV2, CaV3.3, and auxiliary subunits containing plasmids; Dr. Emmanuel Bourinet for kindly donating the CaV3.2-GFP clone (Institut de Génomique Fonctionnelle, Université de Montpellier, Montpellier, France); and Dr. Kees Jalink (Swammerdam Institute for Life Sciences, University of Amsterdam, Amsterdam, The Netherlands) for providing the mTurquoise2-EPAC-cp173Venus-Venus construct.
Funding
This work was supported by grants from the Fondo para la Investigación Científica y Tecnológica (FONCyT, PICT2017-0602 and PICT2019-0460 to JR and PICT2017-3196, PICT2019-3054, and PICT2020-3270 to MP) and from the Universidad Nacional de La Plata (X860 to JR) and the National Qatar Research Foundation (NPRP13S-0209–200315). CIM, ERM, MPC, AY, MP, and JR were supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET). SSR was supported by the Comité de Investigaciones Científicas de Buenos Aires (CICPBA).
Author information
Authors and Affiliations
Contributions
JR directed the conception and experimental design, contributed to data interpretation, and was responsible for funding acquisition. SSR amplified and purified the clones for the HEK293T cells transient transfections. CIM, ERM, and MPC performed data acquisition, analysis, and interpretation. AY performed the cAMP measurements. JR and CIM prepared, wrote, and revised the manuscript. All authors have read, critically revised, and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics Approval
All experiments in this study received approval from the Institutional Animal Care and Use Committee of the IMBICE (approval ID 120319), in strict accordance with the recommendations of the U.S. Guide for the Care and Use of Laboratory Animals of the National Research Council.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
McCarthy, C.I., Mustafá, E.R., Cornejo, M.P. et al. Chlorpromazine, an Inverse Agonist of D1R-Like, Differentially Targets Voltage-Gated Calcium Channel (CaV) Subtypes in mPFC Neurons. Mol Neurobiol 60, 2644–2660 (2023). https://doi.org/10.1007/s12035-023-03221-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03221-1