
Abstract
Mutations of KRAS are found in a variety of human malignancies, including in pancreatic cancer, colorectal cancer, and non-small cell lung cancer at high frequency. To date, no effective treatments that target mutant variants of KRAS have been introduced into clinical practice. In recent years, a number of studies have shown that the oncogene KRAS plays a critical role in controlling cancer metabolism by orchestrating multiple metabolic changes. One of the metabolic hallmarks of malignant tumor cells is their dependency on aerobic glycolysis, known as the Warburg effect. The role of KRAS signaling in the regulation of aerobic glycolysis has been reported in several types of cancer. KRAS-driven cancers are characterized by altered metabolic pathways involving enhanced nutrients uptake, enhanced glycolysis, enhanced glutaminolysis, and elevated synthesis of fatty acids and nucleotides. However, Just how mutated KRAS can coordinate the metabolic shift to promote tumor growth and whether specific metabolic pathways are essential for the tumorigenesis of KRAS-driven cancers are questions which remain to be answered. In this context, the aim of this review is to summarize current data on KRAS-related metabolic alterations in cancer cells. Given that cancer cells rely on changes in metabolism to support their growth and survival, the targeting of metabolic processes may be a potential strategy for treating KRAS-driven cancers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In recent years, there has been intense interest directed towards understanding the reprogramming of metabolic processes in cancer cells [1,2,3,4]. The first recorded observations in cancer metabolism were made by Otto Warburg in the 1920s, who discovered that cancer cells showed increased glycolysis and lactate production regardless of oxygen availability [5]. Accumulating evidence also indicates that the reprogramming of cancer metabolism is under the control of various oncogenic signals [1].
The KRAS proto-oncogene encodes an approximately 21-kDa small GTPase, which cycles between the active guanosine triphosphate-bound state and the inactive guanosine diphosphate-bound state. Oncogenic activation of KRAS can influence several cellular processes that regulate morphology, proliferation, motility, and survival through the activation of its downstream pathways, such as the MAPK and PI3K/AKT/mTOR pathways [6, 7]. KRAS mutations occur in a variety of human malignancies, but they appear most frequently in pancreatic ductal cell carcinoma (PDCA), colorectal cancer (CRC), and non-small cell lung cancer (NSCLC). KRAS-driven cancers are largely resistant to therapeutic intervention, and KRAS itself has been considered to be “undruggable.” To satisfy the increased needs for cellular building blocks as a result of enhanced tumor growth, metabolic pathways are rewired to divert nutrients, such as glucose and glutamine, into anabolic pathways [1]. While most cancers depend on a high rate of aerobic glycolysis for their growth, some cancer cells also display an addiction to glutamine despite glutamine being a non-essential amino acid that can be synthesized from glucose [8,9,10]. Recent studies have shown that oncogenic KRAS promotes metabolic reprogramming through the stimulation of glucose metabolism, differential channeling of glucose intermediates, reprogrammed glutamine metabolism, increased autophagy, and macropinocytosis [11,12,13,14]. The mechanism by which oncogenic KRAS coordinates the shift in metabolism to promote tumor growth remains an area of active investigation.
Autophagy is a highly conserved mechanism to degrade intracellular components and promote cell survival by providing energy in the form of ATP and building blocks (i.e., amino acids, lipids, sugars, and nucleotides) [15]. Autophagy is triggered by nutrient shortage, protein damage, and oxidative stress occurring through the inhibition of the AMP kinase and the mammalian target of rapamycin (mTOR) pathways and the activation of the unfolded protein response (UPR) system. To fuel metabolic processes, KRAS signaling leads to the scavenging of extracellular proteins and lipids, while also activating self-eating and protein recycling processes via autophagy. The role of autophagy in cancer is extremely complex.
Cancer cells are also able to absorb and degrade extracellular components through an endocytic process called macropinocytosis. In PDCA cells, KRAS-dependent upregulation of macropinocytosis acts as an important supply route for amino acids such as glutamine, with macropinocytosis inhibition shown to reduce KRAS-transformed cell growth [16]. Hydroxychloroquine is a compound approved for the treatment of malaria and several rheumatologic diseases that prevents lysosome acidification, thus inhibiting autophagy and macropinocytosis. Hydroxychloroquine is currently being tested in several ongoing trials involving patients with PDCA.
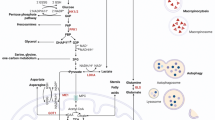
Here, we present a comprehensive review of the metabolic deregulations contributing to KRAS-driven cancer progression (Fig. 1; Table 1). These data reveal several key points that are of prime relevance to KRAS-driven cancers and tumor biology in general. Targeting distinct metabolic features of KRAS-driven cancers provides novel approaches for cancer treatment.

Metabolic alterations in KRAS-driven cancers. Schematic representation of the metabolic routes involved in KRAS-driven cancers. Black boxes Cancer type in which the molecule is associated with mutated KRAS: PDCA pancreatic ductal cell carcinoma, CRC colorectal cancer, NSCLC non-small cell lung cancer. Red arrows Change in expression level that is modulated by mutated KRAS: upwards arrow upregulation, downwards arrow downregulation. GLUT1 Glucose transporter 1, HK1/2 hexokinase 1/2, PFK1 phosphofructokinase 1, GAPDH glyceraldehyde 3-phosphate dehydrogenase, PKM2 pyruvate kinase M2, LDHA lactate dehydrogenase A, GFPT1 glucosamine-fructose-6-phosphate aminotransferase-1, GlcN glucosamine, GlcNAc N-acetylglucosamine, RPE ribulose-5-phosphate-3-epimerase, RPIA ribulose-5-phosphate isomerase, GLS glutaminase, GLUD1 glutamate dehydrogenase 1, GOT glutamate–oxaloacetate transaminase, MDH1 malate dehydrogenase 1, ME1 malic enzyme 1, ASNS asparagine synthetase, α-KG α-ketoglutarate, OAA oxaloacetate, Ac-CoA Acetyl-CoA, FA fatty acid, FASN fatty acid synthase, ACS Acyl-CoA synthetase, HBP hexosamine biosynthesis pathway, PPP pentose phosphate pathway, PDCA pancreatic ductal cell carcinoma, CRC colorectal cancer, NSCLC non-small cell lung cancer
Pancreatic ductal cell adenocarcinoma (PDCA)
PDCA harbors a particular poor prognosis, with a 5-year survival rate of <5% [17]. The current systemic treatment of PDCA is dependent on chemotherapy, with targeted approaches having minimal success. Malignant progression from pancreatic intraepithelial neoplasia to invasive disease is accompanied by an early acquisition of KRAS mutations, which occurs in >90% of cases, and a subsequent loss of tumor suppressors such as INK4A, TP53, and SMAD4.
Glucose metabolism
Studies using the inducible KRAS G12D-driven PDCA mouse model have been very informative for determining which aspects of tumor metabolism are most essential for KRAS-driven cancers. Using this mouse model, Ying et al. reported that mutated KRAS enhances the expression of glucose transporter-1 (GLUT1) and several rate-limiting glycolytic enzymes, including hexokinase and lactate dehydrogenase, and that mutated KRAS maintains tumor growth by stimulating glucose uptake and channeling glucose intermediates into the hexosamine biosynthesis pathway (HBP) and non-oxidative pentose phosphate pathway (PPP): KRAS mutations promote protein glycosylation through HBP and ribose production through non-oxidative PPP [18]. Notably, knockdown of either the HBP gene (Gfpt1) or non-oxidative PPP genes (Rpia or Rpe) lead to inhibition of KRAS-dependent tumor growth in vivo, indicating their potential as therapeutic targets.
Amino acid metabolism
In addition to glucose, glutamine is also an important source of fuel for cancers. Glutamine is the most abundant circulating free amino acid in human plasma. It serves as a major anaplerotic substrate for the tricarboxylic acid (TCA) cycle, and also supplies nitrogen for nucleotides, non-essential amino acids, and hexosamine biosynthesis to fuel cell proliferation. Once transported into the cells via the glutamine transporter (SLC1A5/ASCT2), glutamine is first converted to glutamate by glutaminase (GLS) and then converted to the TCA cycle intermediate, α-ketoglutarate, by glutamate dehydrogenase 1 (GLUD1) or aminotransferases. The glutamine-derived α-ketoglutarate replenishes the TCA cycle by providing oxaloacetate that condenses with acetyl-CoA to maintain the TCA cycle and support fatty acid (FA) biosynthesis. In addition to providing carbon and nitrogen molecules for biosynthesis, glutamine plays a role in the uptake of essential amino acids and in maintaining mTOR signaling and NADPH production for redox control. mTOR complex 1 positively regulates GLS and glutamine flux through the S6K1 (p70 ribosomal S6 kinase 1)-dependent regulation of MYC [19]. The spectrum of glutamine-dependent tumors and the mechanisms by which glutamine supports cancer metabolism are being actively investigated [10, 20, 21]. Son et al. recently reported that KRAS mutations in PDCA regulate glutamine metabolism through its conversion to aspartate, thereby supporting growth by maintaining the cellular redox balance in the PDCA mouse model [22]. While most cells utilize GLUD1 to convert glutamine-derived glutamate into α-ketoglutarate to fuel the TCA cycle, PDCA cells metabolize glutamine through an unconventional pathway in which glutamine is converted to non-essential amino acids, such as aspartate by glutamate–oxaloacetate transaminase 2 (GOT2). Glutamine-derived aspartate is converted into oxaloacetate by aspartate transaminase (GOT1) in the cytoplasm, then converted into malate, and finally converted into pyruvate, resulting in the production of NADPH to maintain the cellular redox balance. In the PDCA mouse model, mutated KRAS affects the reprogramming of glutamine metabolism by downregulating GLUD1 and upregulating GOT1. Importantly, interfering with glutamine metabolism by blocking aspartate transaminase or enzymes downstream could suppress tumor growth of KRAS-driven PDCA. Regarding the glutamine metabolism in PDCA, Wang et al. have reported that arginine methylation at R248 of malate dehydrogenase 1 (MDH1), which is catalyzed by CARM1, regulates glutamine metabolism and redox homeostasis of PDCA cells [23]. Importantly, R248 methylation of MDH1 is observed to be downregulated in clinical PDCA samples.
The increased requirement for amino acids is a very early phenomenon during tumor development. Mayers et al. reported that metabolic reprogramming to provide cancer cells with branched-chain amino acids (BCAAs) preceded PDCA diagnosis by about 5 years and that elevated plasma levels of three BCAAs (isoleucine, leucine, and valine) were associated with future diagnosis of PDCA [24]. They also recently reported that KRAS-driven NSCLC depended on BCAA metabolism, whereas KRAS-driven PDCA did not, indicating the tissue of origin can affect the metabolic dependencies of tumors driven by the same genetic events [25]. NSCLC tumors have an increased expression of the BCAA transporter (Slc7a5) as well as BCAA catabolic enzymes (Bcat2 and Bckdh), which enables BCAAs to be utilized as a nitrogen source, whereas PDCA tumors exhibit decreased expression of these genes.
Autophagy
Autophagy is a highly conserved mechanism which degrades intracellular components and promotes cell survival under metabolic stress by providing energy in the form of ATP and building blocks such as amino acids, lipids, sugars, and nucleotides [15]. While it appears that PDCA cells depend on autophagy for growth, the relationship between oncogenic KRAS and autophagy remains unclear. Genetic or pharmacological inhibition of autophagy results in increased levels of reactive oxygen species (ROS), elevated DNA damage, and mitochondrial defects that have been shown to lead to decreased proliferation of PDCA cell lines in vitro as well as substantial tumor regression in vivo in PDCA mouse models [26, 27]. A recent study with a panel of 47 different cancer cell lines indicated that the KRAS-mutant cells used were no more dependent on autophagy than their wild-type counterparts [28]. Therefore, the specific role of KRAS mutations on autophagy remains controversial in PDCA. Clinical trials are under way in various cancers to test whether inhibiting both autophagy and macropinocytosis can compromise tumor growth by hydroxychloroquine, an inhibitor of lysosomal function. There have been some early reports on the efficacy of hydroxychloroquine in various types of cancer, but the results have been mixed [29,30,31,32].
Macropinocytosis
More recently, it has become apparent that KRAS-driven cancers acquire additional means to satisfy their nutritional needs, such as through fluid-phase endocytic uptake by macropinocytosis and self-cannibalization by autophagy. PDCA cells specifically harboring mutated KRAS utilize macropinocyotsis to transport extracellular protein into the cell [16] where it is used as a source of essential amino acids to sustain cell survival and proliferation [33]. The uptake of serum albumin by macropinocytosis provides amino acids, particularly glutamine, for multiple metabolic pathways, including TCA cycle anaplerosis and macromolecular synthesis. In addition to albumin, RAS-transformed mammalian cells take up exogenous lipids to provide cells with FAs, thereby decreasing the need for de novo synthesis [34]. Preclinical studies in which heterotopic xenografts-bearing mice were treated with a compound 5-(N-ethyl-N-isopropyl) amiloride, an inhibitor of macropinocytosis, showed an attenuation of tumor growth [16].
Colorectal cancer (CRC)
Carcinogenesis of CRC is caused by genetic mutations in various genes, such as APC, KRAS, p53, SMAD4, and PTEN, as well as by the epigenetic silencing of tumor suppressor genes [35]. KRAS mutations occur in approximately 40% of CRCs, and a number of studies have shown that KRAS mutations in CRC predict a lack of responses to therapies with antibodies targeting the epidermal growth factor receptor (EGFR) [36, 37]. Cetuximab and panitumumab, which are anti-EGFR monoclonal antibodies, are now recommended only for patients whose tumors have wild-type KRAS.
The international CRC Subtyping Consortium shares 18 large-scale data sets and has revealed that CRCs can be classified into four consensus molecular subtypes (CMSs), each with distinct features: CMS1 (hypermutated, microsatellite instability, and strong immune activation), CMS2 (epithelial, WNT and MYC activation), CMS3 (metabolic dysregulation) and CMS4 (transforming growth factor beta activation, stromal invasion, and angiogenesis) [38]. Notably, CMS3 is characterized by prominent metabolic dysregulation and is strongly correlated with KRAS mutations, which may suggest that the targeted intervention to the metabolic abnormalities is especially promising for KRAS-mutant CRCs.
Glucose metabolism
There are only a few reports on KRAS-related metabolic alterations in CRCs. Yun et al. reported particularly interesting data showing that the increase in GLUT1 expression and glucose uptake was critically dependent on KRAS and BRAF mutations in CRC cell lines and that this metabolic alteration provided a distinct survival advantage because CRC cells with mutated KRAS or BRAF were able to survive long-term in low-glucose culture environments [39]. In CRC cells with mutated KRAS or BRAF, increased glucose transport was associated with increased lactate production, although mitochondrial function and oxidative respiration were not affected [39]. Importantly, 3-bromopyruvate (3-BrPA), a glycolysis inhibitor, was highly effective on the xenografts that were derived from CRC cells with mutated KRAS or BRAF [39]. These results provide a proof of principle that glycolytic inhibitors can retard tumor growth at doses that are non-toxic to normal tissues.
Positron emission tomography with 18F-fluorodeoxyglucose (FDG) is a diagnostic tool routinely used to detect cancers in the clinical setting. Using this technique clinicians are able to evaluate glucose metabolism in vivo by measuring the uptake of FDG, a glucose analog. Although FDG accumulation in tumor cells largely depends on GLUT1 and the rate-limiting glycolytic enzyme hexokinase type-2 (HK2), several recent studies on CRCs have reported that increased GLUT1 expression is the most essential factor for FDG accumulation [40]. In a retrospective study with primary and metastatic CRCs, we previously reported that FDG accumulation by KRAS-mutant CRCs was significantly higher than that by CRCs with wild-type KRAS [41,42,43]. There is also emerging evidence from other groups that FDG accumulation reflects the mutational status of KRAS in CRC and NSCLC [44,45,46,47]. In terms of the underlying mechanisms behind these clinical observations, we have recently shown that mutated KRAS causes higher FDG accumulation, possibly by upregulating GLUT1 and at least partially by upregulating hypoxia-inducible factor 1-alpha (HIF-1α) induction under hypoxic condition [48].
Yun et al. recently reported that high levels of vitamin C were selectively toxic to CRCs with mutated KRAS or BRAF because the increased uptake of oxidized vitamin C via elevated GLUT1 expression disrupted redox homeostasis by depleting glutathione [49]. Accumulation of ROS mediated by an increased uptake of the oxidized vitamin C inhibited glycolysis at the level of glyceraldehyde 3-phosphate dehydrogenase, which led to an energetic crisis and ultimately cell death [49]. Regarding the antitumoral mechanism of vitamin C in KRAS-mutant CRC, Aguilera et al. have recently reported that vitamin C interferes with the downstream RAS/ERK pathway by facilitating the detachment of KRAS protein from the cell membrane, and then downregulates expression of GLUT1 and pyruvate kinase M2, which results in an energetic crisis [50]. Overall, these results provide a mechanistic rationale for the therapeutic use of vitamin C for KRAS-mutant CRCs.
Amino acid metabolism
Our group recently performed a comprehensive metabolomics analysis of isogenic CRC cell lines harboring mutated KRAS and wild-type KRAS and observed that KRAS mutations in CRC can cause alterations in amino acid metabolism [51]. These alterations are especially prominent in glutamine metabolism, where a marked decrease in aspartate level and an increase in asparagine level were observed [51]. We also found that asparagine synthetase (ASNS), the enzyme that synthesizes asparagine de novo from aspartate, was upregulated by the KRAS-activated signaling pathway, in particular by the PI3/K-AKT/mTOR pathway, and that KRAS-mutant CRC cells could become adaptive to glutamine depletion through asparagine biosynthesis synthesized via ASNS. Importantly, tumor growth in vivo of KRAS-mutant CRC cells was significantly suppressed upon ASNS knockdown, indicating that ASNS could be a novel therapeutic target. We also observed that mutated KRAS did not alter the expression of GLUD1 or GOT1 in CRC, although mutated KRAS has been reported to cause a decrease in GLUD1 and an increase in GOT1 in PDCA [22], which indicates the multifaceted roles of mutated KRAS in metabolism are cell type-dependent. In human glioma and neuroblastoma, asparagine plays a critical role in regulating cellular adaptation to glutamine depletion [52]. Asparagine is an essential component to suppress glutamine-withdrawal-induced apoptosis without restoring other non-essential amino acids or TCA cycle intermediates, and ASNS expression is statistically correlated with poor prognosis of glioma and neuroblastoma patients. Hettner et al. recently reported that ASNS silencing had the strongest inhibitory effect on sarcoma growth in a functional genomic short hairpin RNA (shRNA)-based screening of genetically engineered mouse sarcoma generated by oncogenic KRAS and disruption of Cdkn2a [53]. These authors also observed that ASNS inhibition significantly inhibited sarcoma growth in vivo only when combined with the depletion of plasma asparagine, which indicates that asparagine can promote cellular adaptation to metabolic stress such as glutamine depletion. ASNS is activated by mutated p53, protein limitation, and tumor microenvironmental stress [54, 55]. Although normal pancreatic tissues express high levels of ASNS, approximately half of PDCA express no ASNS or only low ASNS levels. These tumors may harbor an intrinsic fragility to asparagine depletion that could be exploited by L-asparaginase therapy [56]. Potent ASNS inhibitors has also been developed as new drugs for L-asparaginase-resistant acute lymphoblastic leukemia [57, 58]. The addition of asparagine to glutamine-deprived cells alters the transcriptional response, thereby suppressing the induction of apoptotic regulators of the UPR effectors CHOP and XBP1 [59]. Furthermore, a recent report indicates that intracellular asparagine promotes cancer cell proliferation as an exchange factor of extracellular amino acids (serine, arginine, and histidine), which is involved in the synthesis of proteins and nucleotides [59]. Taken together, these results suggest that asparagine may play a central role as an important regulator of cancer cell amino acid homeostasis, anabolic metabolism, and cell proliferation.
There is also emerging evidence from other research groups that KRAS mutations in CRC are associated with glutamine metabolism. Weinberg et al. reported that PPP, not glycolysis, was essential for KRAS-induced CRC cell growth under the aerobic condition and that glutamine conversion into the TCA cycle intermediate α-ketoglutarate via glutaminase and alanine aminotransferase was essential for KRAS-induced anchorage-independent growth in vitro [60]. Wong et al. have recently reported that CRC cells harboring mutations in KRAS and APC/CTNNB1 are selectively sensitive to knockdown of the mitochondrial glutamine transporter SLC25A22 and that he knockdown of SLC25A22 suppresses glutaminolysis and aspartate biosynthesis via the TCA cycle, leading to apoptosis and cell cycle arrest [61]. In terms of its clinical significance, SLC25A22 overexpression is associated with poor prognosis in patients harboring KRAS mutations. Miyo et al. have very recently reported that the resistance to glucose-deprived conditions in CRC is associated with the levels of GLUD1 and SLC25A13 (a mitochondrial aspartate–glutamate carrier) and that combined expression of GLUD1 and SLC25A13 is significantly associated with tumor aggressiveness and poorer prognosis of patients with CRC [62]. Previous studies have demonstrated that mutated KRAS controls the reprogramming of glutamine metabolism by decreasing GLUD1 and increasing GOT1 in the PDCA mouse model [22]. An important question remains as to whether these novel glutamine pathways are active in all KRAS-mutant tumors or if they are specific to PDCAs with KRAS mutations. Recent data on CRCs from our group [51] and other research groups [61, 62] suggest the latter whereby this specificity may not be a global property of all tumor types harboring KRAS mutations. The role of glutamine transporters, such as ASCT2 and LAT1, in cancer is also under investigation as part of the new era in the discovery of novel anticancer drugs [63, 64].
Non-small cell lung cancer (NSCLC)
NSCLC is a leading cause of cancer-related deaths worldwide, and KRAS mutations occur in approximately 30% of NSCLC [65]. In the vast majority of cases, KRAS mutations for EGFR or ALK are found in wild-type tumors; in other words, these mutations are non-overlapping with other oncogenic mutations found in NSCLC. Therefore, KRAS mutations define a distinct molecular subset of the disease. Unlike in CRC, KRAS mutations have not yet been shown in NSCLC to be negative predictors of a benefit of the anti-EGFR therapy. Recently, a systematic approach with a pool-based shRNA screening revealed that a combination of trametinib, a MEK inhibitor, plus fibroblast growth factor receptor (FGFR) inhibitor could be a promising strategy for treating KRAS-mutant NSCLC [66].
Glucose metabolism
Hexokinases catalyze the first committed step of glucose metabolism. HK2 was identified as an attractive target for KRAS-driven NSCLC and ErbB2-driven breast cancer because systemic whole-body deletion of HK2 in these mouse models inhibited tumor initiation and maintenance [67]. HK2 was highly expressed in cancer cells and not in the normal cells, thus allowing for selective targeting of cancer cells. HK2 deletion in KRAS-driven NSCLC cells suppressed glucose-derived ribonucleotide synthesis and impaired the incorporation of glutamine-derived carbon into TCA cycle intermediates [67].
Amino acid metabolism
Weinberg et al. reported that glutamine conversion into the TCA cycle intermediate α-ketoglutarate allowed the generation of ROS, which was required for KRAS-induced tumorigenicity through the ERK-MAPK pathway in the KRAS-driven NSCLC mouse model [60]. The role of other amino acids, such as serine and glycine, in the metabolism of KRAS-driven cancers remains to be investigated, although serine and glycine metabolism has recently been reported to play a key role in cancer cell proliferation and survival [68, 69].
Autophagy
In a mouse model of spontaneous KRAS G12D-driven NSCLC, knockout of Atg7, an essential autophagy gene, caused the accumulation of dysfunctional mitochondria, suppressed tumor growth, and diverted the progression of carcinomas to more benign oncocytomas [70]. Autophagy deficiency in KRAS G12D-driven NSCLC cells enhanced the dependence on glutamine, indicating that protein degradation by autophagy supplies amino acids and their derivatives to metabolic pathways, of which glutamine is partially critical [70].
Lipogenesis
Fatty acids are fundamental cellular components used for post-translational protein modification and energy generation through β-oxydation. De novo FA synthesis involves several enzymes: ATP citrate lyase generates acetyl-CoA from citrate; acetyl-CoA carboxylase catalyzes carboxylation of acetyl-CoA to form malonyl-CoA; fatty acid synthase (FASN) adds 2-carbon units to form long-chain FAs; acyl-CoA synthetase (ACS) converts long-chain FAs into acyl-CoA. The metabolism of FAs is emerging as a mechanism to cope with KRAS mutations. For example, mutated KRAS stimulates scavenging unsaturated FAs from lysophospholipids under hypoxic conditions in mammalian cells [34]. Padanad et al. have recently reported that mutated KRAS regulates lipid biosynthesis through ACS long-chain family member 3 (ACSL3) and that ACSL3 promotes cellular uptake, accumulation and β-oxydation of FAs, which is required for lung tumorigenesis in the KRAS-driven NSCLC mouse model [71]. Gouw et al. have recently reported that mutated KRAS activates lipogenesis through induction of FASN, which results in ERK2 activation and lipid signatures associated with human lung cancer cell lines [72].
Conclusions
Since cancer cells have distinct metabolic requirements that differ from those of their normal counterparts, they may be more reliant on specific fuel sources, thus providing unique therapeutic targets. However, there is still much work to be done to fully realize the potential of these approaches. It is likely that KRAS mutations have tissue-specific effects on metabolism due to the intrinsic metabolic wiring in the tissue of origin of a particular tumor. It is also important to incorporate the tumor suppressor background when studying the effects of KRAS mutations on metabolism. In addition, more studies are required to investigate how these oncogenic KRAS-dependent metabolic changes are altered in vivo in the tumor microenvironment, such as hypoxia, limited nutrients, and crosstalk between tumor cells and stromal cells. Davidson et al. recently reported that KRAS-driven NSCLC cells in vitro use nutrients differently than lung tumors in vivo, especially with regard to glutamine metabolism; KRAS-driven lung tumors were less dependent on glutaminase than cultured cells [73]. This difference highlights the importance of studying cancer metabolism in a physiological context. About 20% of human tumors have mutations in RAS, most frequently in KRAS (85%), NRAS (15%), and HRAS (1%). Although about 20% of melanoma and about 2–4% of CRC have mutations in NRAS, little is known about the metabolic alterations of mutated NRAS signaling [74, 75].
In conclusion, we have summarized and highlighted the recently defined role of oncogenic KRAS in the regulation of altered metabolic signaling pathways in KRAS-driven cancers.
Abbreviations
- ACS:
-
Acyl-CoA synthetase
- ASNS:
-
Asparagine synthetase
- BCAA:
-
Branched-chain amino acid
- CMS:
-
Consensus molecular subtype
- CRC:
-
Colorectal cancer
- EGFR:
-
Epidermal growth factor receptor
- FA:
-
Fatty acid
- FASN:
-
Fatty acid synthase
- FDG:
-
Fluorodeoxyglucose
- FGFR:
-
Fibroblast growth factor receptor
- GLS:
-
Glutaminase
- GLUD1:
-
Glutamate dehydrogenase 1
- GLUT1:
-
Glucose transporter-1
- GOT:
-
Glutamate–oxaloacetate transaminase
- HBP:
-
Hexosamine biosynthesis pathway
- HK:
-
Hexokinase
- MDH1:
-
Malate dehydrogenase 1
- mTOR:
-
Mammalian target of rapamycin
- NSCLC:
-
Non-small cell lung cancer
- PDCA:
-
Pancreatic ductal cell carcinoma
- PET:
-
Positron emission tomography
- PKM2:
-
Pyruvate kinase M2
- PPP:
-
Pentose phosphate pathway
- ROS:
-
Reactive oxygen species
- TCA:
-
Tricarboxylic acid
- UPR:
-
Unfolded protein response
References
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 11:325–337
Cairns RA, Harris IS, Mak TW (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11:85–95
Galluzzi L, Kepp O, Vander Heiden MG et al (2013) Metabolic targets for cancer therapy. Nat Rev Drug Discov 12:829–846
Warburg O (1956) Injuring of respiration the origin of cancer cells. Science 123:309–314
Karnoub AE, Weinberg RA (2008) Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 9:517–531
Gysin S, Salt M, Young A et al (2011) Therapeutic strategies for targeting ras proteins. Genes Cancer 2:359–372
Eagle H (1955) Nutrition needs of mammalian cells in tissue culture. Science 122:501–514
Kovacević Z, Morris HP (1972) The role of glutamine in the oxidative metabolism of malignant cells. Cancer Res 32:326–333
Wise DR, Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35:427–433
Bryant KL, Mancias JD, Kimmelman AC et al (2014) KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci 39:91–100
Cohen R, Neuzillet C, Tijeras-Raballand A et al (2015) Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 6:16832–16847
Kimmelman AC (2015) Metabolic dependencies in RAS-driven cancers. Clin Cancer Res 21:1828–1834
White E (2013) Exploiting the bad eating habits of Ras-driven cancers. Genes Dev 27:2065–2071
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330:1344–1348
Commisso C, Davidson SM, Soydaner-Azeloglu RG et al (2013) Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497:633–637
Vincent A, Herman J, Schulick R et al (2011) Pancreatic cancer. Lancet 378:607–620
Ying H, Kimmelman AC, Lyssiotis CA et al (2012) Oncogenic KRAS maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149:656–670
Csibi A, Lee G, Yoon SO et al (2014) The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol 24:2274–2280
DeBerardinis RJ, Cheng T (2010) Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29:313–324
Bhutia YD, Babu E, Ramachandran S et al (2015) Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res 75:1782–1788
Son J, Lyssiotis CA, Ying H et al (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496:101–105
Wang YP, Zhou W, Wang J et al (2016) Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol Cell 64:673–687
Mayers JR, Wu C, Clish CB et al (2014) Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med 20:1193–1198
Mayers JR, Torrence ME, Danai LV et al (2016) Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353:1161–1165
Yang S, Wang X, Contino G et al (2011) Pancreatic cancers require autophagy for tumor growth. Genes Dev 25:717–729
Guo JY, Chen HY, Mathew R et al (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25:460–470
Eng CH, Wang Z, Tkach D et al (2016) Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci USA 113:182–187
White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12:401–410
Rosenfeld MR, Ye X, Supko JG et al (2014) A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10:1359–1368
Mahalingam D, Mita M, Sarantopoulos J et al (2014) Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 10:1403–1414
Wolpin BM, Rubinson DA, Wang X et al (2014) Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 19:637–638
Palm W, Park Y, Wright K et al (2015) The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162:259–270
Kamphorst JJ, Cross JR, Fan J et al (2013) Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci USA 110:8882–8887
Vogelstein B, Kinzler KW (2004) Cancer genes and the pathways they control. Nat Med 10:789–799
Karapetis CS, Khambata-Ford S, Jonker DJ et al (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancers. N Engl J Med 359:1757–1765
Lievre A, Bachet JB, Boige V et al (2008) KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 26:374–379
Guinney J, Dienstmann R, Wang X et al (2015) The consensus molecular subtypes of colorectal cancer. Nat Med 21:1350–1356
Yun J, Rago C, Cheong I et al (2009) Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325:1555–1559
Jadvar H, Alavi A, Gambhir SS (2009) 18F-FDG uptake in lung, breast, and colon cancers: molecular biology correlates and disease characterization. J Nucl Med 50:1820–1827
Kawada K, Nakamoto Y, Kawada M et al (2012) Relationship between 18F-fluorodeoxyglucose accumulation and KRAS/BRAF mutations in colorectal cancer. Clin Cancer Res 18:1696–1703
Kawada K, Toda K, Nakamoto Y et al (2015) Relationship between 18F-FDG PET/CT scans and KRAS mutations in metastatic colorectal cancer. J Nucl Med 56:1322–1327
Kawada K, Iwamoto M, Sakai Y (2016) Mechanisms underlying 18F-fluorodeoxyglucose accumulation in colorectal cancer. World J Radiol 8:880–886
Chen SW, Chiang HC, Chen WT et al (2014) Correlation between PET/CT parameters and KRAS expression in colorectal cancer. Clin Nucl Med 39:685–689
Miles KA, Ganeshan B, Rodriguez-Justo M et al (2014) Multifunctional imaging signature for V-KI-RAS2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations in colorectal cancer. J Nucl Med 55:386–391
Lee JH, Kang J, Baik SH et al (2016) Relationship between 18F-Fluorodeoxyglucose uptake and V-Ki-Ras2 kirsten rat sarcoma viral oncogene homolog mutation in colorectal cancer patients: variability depending on c-reactive protein level. Medicine 95:e2236
Caicedo C, Garcia-Velloso MJ, Lozano MD et al (2014) Role of [1∙F]FDG PET in prediction of KRAS and EGFR mutation status in patients with advanced non-small-cell lung cancer. Eur J Nucl Med Mol Imaging 41:2058–2065
Iwamoto M, Kawada K, Nakamoto Y et al (2014) Regulation of 18F-FDG accumulation in colorectal cancer cells with mutated KRAS. J Nucl Med 55:2038–2044
Yun J, Mullarky E, Lu C et al (2015) Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350:1391–1396
Aguilera O, Muñoz-Sagastibelza M, Torrejón B et al (2016) Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 7:47954–47965
Toda K, Kawada K, Iwamoto M et al (2016) Metabolic alterations caused by KRAS mutations in colorectal cancer contribute to cell adaptation to glutamine depletion by upregulation of asparagine synthetase. Neoplasia 18:654–665
Zhang J, Fan J, Venneti S et al (2014) Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell 56:205–218
Hettmer S, Schinzel AC, Tchessalova D et al (2015) Functional genomic screening reveals asparagine dependence as a metabolic vulnerability in sarcoma. Elife 4:e09436. doi:10.7554/eLife.09436
Ye J, Kumanova M, Hart LS et al (2010) The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 29:2082–2096
Balasubramanian MN, Butterworth EA, Kilberg MS (2013) Asparagine synthetase: regulation by cell stress and involvement in tumor biology. Am J Physiol Endocrinol Metab 304:789–799
Dufour E, Gay F, Aguera K et al (2012) Pancreatic tumor sensitivity to plasma l-asparagine starvation. Pancreas 41:940–948
Richards NG, Kilberg MS (2006) Asparagine synthetase chemotherapy. Annu Rev Biochem 75:629–654
Ikeuchi H, Ahn YM, Otokawa T et al (2012) A sulfoximine-based inhibitor of human asparagine synthetase kills l-asparaginase-resistant leukemia cells. Bioorg Med Chem 20:5915–5927
Kral AS, Xu S, Graeber TG et al (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun 7:11457
Weinberg F, Hamanaka R, Wheaton WW et al (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 107:8788–8793
Wong CC, Qian Y, Li X et al (2016) SLC25A22 promotes proliferation and survival of colorectal cancer cells with KRAS mutations and xenograft tumor progression in mice via intracellular synthesis of aspartate. Gastroenterology 151(945–960):e6
Miyo M, Konno M, Nishida N et al (2016) Metabolic adaptation to nutritional stress in human colorectal cancer. Sci Rep 6:38415
Fuchs BC, Bode BP (2005) Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol 15:254–266
Bhutia YD, Ganapathy V (2016) Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim Biophys Acta 1863:2531–2539
Herbst RS, Heymach JV, Lippman SM (2008) Lung cancer. N Engl J Med 359:1367–1380
Manchado E, Weissmueller S, Morris JP 4th (2016) A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 534:647–651
Patra KC, Wang Q, Bhaskar PT et al (2013) Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24:213–228
Jain M, Nilsson R, Sharma S et al (2012) Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336:1040–1044
Kim D, Fiske BP, Birsoy K et al (2015) SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 520:363–367
Guo JY, Karsli-Uzunbas G, Mathew R et al (2013) Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27:1447–1461
Padanad MS, Konstantinidou G, Venkateswaran N (2016) Fatty acid oxidation mediated by Acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep 16:1614–1628
Gouw AM, Eberlin LS, Margulis K (2017) Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci USA 114:4300–4305
Davidson SM, Papagiannakopoulos T, Olenchzock BA et al (2016) Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab 23:517–528
Zhou B, Der CJ, Cox AD (2016) The role of wild type RAS isoforms in cancer. Semin Cell Dev Biol 58:60–69
Ratnikov BI, Scott DA, Osterman AL (2017) Metabolic rewiring in melanoma. Oncogene 36:147–157
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no potential conflicts of interest to be disclosed.
About this article

Cite this article
Kawada, K., Toda, K. & Sakai, Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int J Clin Oncol 22, 651–659 (2017). https://doi.org/10.1007/s10147-017-1156-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10147-017-1156-4