
Abstract
Cancer cells rewire their metabolism in order to boost growth, survival, proliferation, and chemoresistance. The common event of this aberrant metabolism is the increased glucose uptake and fermentation of glucose to lactate. This phenomenon is observed even in the presence of O2 and completely functioning mitochondria. This is known as the “Warburg Effect” and it is a hallmark in cancer. Up to 40% of all CRC’s are known to have a mutated (abnormal) KRAS gene, found at differing frequencies in all consensus molecular subtypes (CMS). CMS3 colon cancer molecular subtype contains the so-called ‘metabolic tumours’ which represents 13% of total CR cases. These tumours display remarkable metabolic deregulation, often showing KRAS mutations (68%). Unfortunately, patients harbouring mutated KRAS are unlikely to benefit from anti-EGFR therapies. Moreover, it remains unclear that patients with KRAS wild-type CRC will definitely respond to such therapies. Although some clinically designed-strategies to modulate KRAS aberrant activation have been designed, all attempts to target KRAS have failed in the clinical assays and KRAS has been assumed to be invulnerable to chemotherapeutic attack. Quest for metabolic inhibitors with anti-tumour activity may constitute a novel and hopeful approach in order to handle KRAS dependent chemoresistance in colon cancer.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Introduction
According to GLOBOCAN (2012), the estimated number of cancer cases worldwide in 2008 was 12.7 million, with 7.6 million deaths. By 2030, there will an estimated 22.2 million newly diagnosed cancer cases and 12 million deaths (Ferlay et al. 2015).
Colorectal cancer (CRC) is a frequently lethal disease showing diverse outcomes and chemotherapy responses. Currently, CRC is the third leading site of cancer in men and women and is the second leading cause of cancer-related deaths.
This number will increase largely by growth and aging of populations and will be largest in low- and medium-resource countries.
The global distribution of neoplasia and types of cancer with higher penetrance continues to change, especially in economically developing countries. Lower income countries accounted for about half (51%) of all cancers worldwide in 1975 but this proportion increased to 55% in 2007 and it is projected to reach 61% by 2050 (Bray and Møller 2006).
Nowadays, oncology tries to personalize anti-cancer therapy on the basis of tumour genotypes in order to provide enhanced prognostic and treatment planning.
In 2015, the molecular classification carried out by Guinney et al. established four different CRC subtypes with specific molecular features: CMS1 (MSI Immune, 14%), hypermutated, microsatellite unstable, remarkable immune activation; CMS2 (Canonical, 37%), epithelial, chromosomally unstable, marked WNT and MYC signalling activation; CMS3 (Metabolic, 13%), epithelial, discernible metabolic dysregulation; and CMS4 (Mesenchymal, 23%), prominent transforming growth factor β activation, stromal invasion, and angiogenesis.
Despite the frequency of KRAS mutations in cancer patients, data outcome is confusing regarding the impact these mutations have on treatment response and patient outcomes.
Although the development of molecular-targeted therapy has supposed a clear benefit on the survival of patients with metastatic CRC, the majority of patients with stage IV CRC undergoing complete resection die from metastasis (Karapetis et al. 2008; Hurwitz et al. 2004).
CRC tumorigenesis is characterized by the accumulation of sequential genetic and epigenetic alterations, and V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations are an early step in tumorigenesis (Vogelstein et al. 1988; Aguilera et al. 2006).
RAS genes are among the most frequently mutated genes in human cancer. Scientific evidence indicates that mutations activating RAS family members, KRAS, HRAS, and NRAS, are found in 20–30% of all human tumours (Cox et al. 2014; Prior et al. 2012). Although KRAS mutations are present in every molecular subtype defined by Guinney et al. in 2015, they are more prevalent in CMS3 CRC (68%). Interestingly, CMS3 appeared to be the most similar to normal colon tissue at the gene expression level.
Recently, it has been suggested that the precursor lesion to KRAS mutant CRC (the majority of CMS3 cancers) are tubovillous adenomas with serrated traits.
Cetuximab and panitumumab are monoclonal antibodies directed to the exodomain of the epidermal growth factor receptor (EGFR) , which in turn blocks downstream signalling, including the RAS/RAF/MEK/ERK pathway. Cetuximab and panitumumab have been proven to be effective in patients with metastatic colorectal cancer (mCRC) when administered alone or in combination with standard chemotherapy (Jonker et al. 2007; van Cutsem et al. 2009).
Cetuximab, is commonly used in the clinical practice to treat metastatic colon, and head and neck cancer. The U.S. Food and Drug Administration (FDA) approved Panitumumab (INN), formerly ABX-EGF, in 2006 for “the treatment of EGFR -expressing metastatic colorectal cancer with disease progression”. However, benefits of such therapies in the survival outcome of patients harbouring KRAS mutant CRC are dramatically decreased.
Here we aim to describe different molecular approaches focused on overcoming the molecular barriers posed by KRAS mutation, often shown in metabolic CMS3 tumours, targeting key players involved in the metabolic reprogramming in colorectal tumours.
3.2 Reprogramming Cell Metabolism in Cancer: The “Warburg Effect”
After almost a century ago since the Warburg effect was described in 1924, this atypical metabolism is considered a hallmark in every type of cancer, exhibiting higher glycolysis and lactate metabolism and defective mitochondrial ATP production.
Although cancer is a heterogeneous malady, often considered as different diseases converging in the abnormal cell growth, cancer cells share the very same metabolic trait: abnormal rates of glucose conversion to lactate even in the presence of O2 (Warburg 1956).
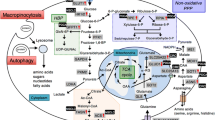
In general, cancer cells metabolize glucose, lactate, hydroxybutyrate, pyruvate, lactate, fatty acids and glutamine at much higher rates than normal ones. In spite of the higher glycolytic rates often shown by cancer cells, ATP rendering is substantially lower than normal cells. Unlike, molecular biosynthesis requirements of tumour cells are markedly higher (Fig. 3.1a, b).

Targeting metabolism in colon cancer. (a) In normal tissues, cell may obtain energy through Oxidative Phosphorylation (OxPhos) that generates 36 ATP. In poor oxygen conditions, normal tissues may obtain energy via anaerobic glycolysis, which gives 2 ATP. For the same glucose, normal cells will get 18 times more energy using oxygen in the mitochondrion compared to anaerobic glycolysis. Normal tissues only use this less efficient pathway in the absence of oxygen. In cancer cells the situation changes dramatically. Even in the presence of oxygen, cancer cells use a less efficient method of energy generation (glycolysis, not phosphorylation) delivering only 4 ATP per molecule of glucose. (b) Unlike cancer cells, in normal cells the main goal for glucose uptake is producing enough ATP to keep cell metabolic homeostasis. (c) Described molecules with antitumor activity targeting metabolism in colon cancer
However, the metabolic homeostasis of tumours is complex because they contain multiple niches, which are linked by the transfer of these catabolites. This metabolic plasticity enables cancer cells to produce ATP, while keeping the reduction–oxidation (redox) balance and committing resources to biosynthesis processes that are essential for cell survival, growth, and proliferation (Martinez-Outschoorn et al. 2017).
This metabolic feature, known as the “Warburg effect”, represents in fact the basis for the 18F-fluorodeoxyglucose positron emission tomography (FDG-PET), a non- invasive imaging technique that, providing an accurate assessment of tumour glucose utilization, is widely exploited in the clinic for initial diagnosis, measuring tumour size, staging, and monitoring tumour responses to therapies. The Warburg effect allows promoting the deviation of glycolytic metabolites into multiple correlated pathways that provide substrates for the biosynthesis of macromolecules (lipids, nucleic acids, and proteins) required for rapid tumour growth and proliferation.
Furthermore, an augmented flux of cytosolic NADPH provided from the cytosolic oxidative pentose phosphate pathway (PPP) is observed that will balance the intracellular redox potential and neutralize the excessive levels of reactive oxygen species (ROS) resulting from the enhanced metabolic activity of cancer cells.
Warburg hypothesized that cancer was caused by defects in mitochondrial oxidative phosphorylation and then forcing the cell to switch into glycolysis, thus cells would become undifferentiated and cancerous. However, studies carried out by Prof. Craig B . Thompson’s laboratory at the Memorial Sloan-Kettering Cancer Center, preferentially indicates that the Warburg effect is not just a passive response to damaged mitochondria but results from oncogene -directed metabolic reprogramming required to support glycolytic metabolism and anabolic growth (Ward and Thompson 2012). The question about altered metabolism as primary cause or just consequence of genomic alteration in cancer still remains open.
Nevertheless, alterations in oncogenic signal transduction pathways and loss of tumour suppressor genes , then affecting the regulation of enzymes and transporters, are tightly correlated to aerobic glycolysis in cancer. Over-expression of the facilitative glucose transporter 1 (GLUT-1) promotes an increased glucose uptake in a variety of cancer cell types, and has been associated with the loss of the tumour suppressor PTEN (Morani et al. 2014) or constitutive activation of oncogenes such as KRAS, AKT, SRC, and Myc (Dang 2012).
HIF-1 and Myc positively regulate the expression of pyruvate kinase M2 (PKM2), the final rate-limiting enzyme of glycolysis, which catalyses the conversion of phosphoenolpyruvate to pyruvate.
Aberrant activity of some signalling pathways such as the PI3K /AKT/mTORC1 pathway has been reported to enhance the expression of glycolytic enzyme Hexokinase II (HKII) that mediates the phosphorylation of glucose upon its entrance into the cell (Wang et al. 2014).
HIF-1 and c-Myc are also considered master regulators of the metabolic reprogramming in neoplasia. Both proteins have been shown to cooperatively induce the expression of both, HKII and pyruvate dehydrogenase kinase 1 (PDHK1). PDHK1 in turn inactivates the pyruvate dehydrogenase (PDH) and so the pyruvate dehydrogenase enzyme complex (PDC) involved in the conversion of pyruvate to acetyl-coenzyme A, therefore inhibiting pyruvate entrance in the tricarboxylic acid (TCA) cycle and then limiting mitochondrial OXPHOS.
Moreover, both HIF-1 and c-Myc enhance the transcription of lactate dehydrogenase A (LDHA), which catalyses the transformation of pyruvate into lactate, further then boosting the glycolytic cancer phenotype in Myc over- expressing cancers (Dang et al. 2008).
Over-expression of PKM2 has been found in multiple human cancers, including colon cancer, and it is considered a master regulator of the metabolic rewiring (Aguilera et al. 2016).
PKM2 may form a complex tetramer, with high catalytic activity, or can exist as a dimer, less active form. In cancer cells, different post-translational modifications, such as fibroblast growth factor receptor type 1 (FGFR1)-mediated phosphorylation at Tyr105, enhance the tetramer-to-dimer switch, therefore inhibiting pyruvate kinase activity and promoting the diversion of glycolytic intermediates towards collateral pathways, such as PPP and serine biosynthesis (Dayton et al. 2016).
3.3 Mutant KRAS in Colon Cancer
Genetic alterations in any one of the 3 isoforms of the RAS family (HRAS, NRAS, or KRAS genes) are very frequent events on neoplasic transformation. The Sanger Centre keeps and periodically updates an exhaustive database involving the nature and frequency of RAS mutations in different human tumours (catalogue of somatic mutations in cancer: http://sanger.ac.uk/cosmic).
In fact, activating KRAS mutations are found in human epithelial neoplasia with an overall frequency of 30%. Studies of the extensive panels of human tumour tissue samples analysed during the last three decades have shown that there is a prevalent correlation of specific mutated RAS isoforms with particular types of tumours (Shimizu et al. 2007).
Some reports describing KRAS overexpression in a colon carcinoma did not find a positive correlation between KRAS overexpression and prognosis, pointing out that RAS overexpression should not be used as a predictive factor (Akkiprik et al. 2008).
KRAS mutations are detected in 40–45% of all colorectal carcinoma (CRC) suggesting that KRAS proteins must play a pivotal role in tumour development (Vaughn et al. 2011). Most KRAS mutations are located in codons 12 and 13 (80% and 20%, respectively), being G12D the most often amino acid modification. On the other hand, much lower activating mutation percentages have been found in NRAS (1–3%) (Irahara et al. 2010). KRAS mutation is an early event in CRC . Although still arguable, it has been proposed that in some CRCs, KRAS mutations may occur as early events in formation of aberrant crypt foci that could later trigger the development of hyperplasic polyps and eventually to CRC (Yuen et al. 2002; Nash et al. 2010). However, contrary to pancreatic carcinomas where KRAS mutations are prevalent, many other genetic and epigenetic alterations besides KRAS mutations may occur in CRC that could be responsible for tumour initiation and progression in this case like APC , beta-catenin mutations and promoter silencing of genes involved in controlling the WNT signalling pathway.
As it has been reported on pancreatic ductal adenocarcinoma (PDAC), there is a tight correlation between KRAS mutations and poor prognosis of aggressive and invasive colorectal carcinomas (Conlin et al. 2005; Zavodna et al. 2009). Interestingly, some clinical studies have reported that the rate of KRAS mutation is higher in CRC patients with lung metastasis and that the presence of the mutation in CRC patients with liver metastasis correlates with poor prognosis (Cejas et al. 2009; Nash et al. 2010).
3.4 Metabolic Switch in CRC: KRAS Connection
As it has been widely described, several clinical trials have shown that KRAS mutations in cancer may predict a lack of responses to the anti-epidermal growth factor receptor (EGFR)–based therapy. So, anti-EGFR therapies using cetuximab and/or panitumumab are now limited to patients with KRAS wild-type CRC (Jonker et al. 2007; Karapetis et al. 2008; Ye et al. 2013).
Interestingly, mutant KRAS is closely involved in the upregulation of the cell Warburg metabolism. Mutated KRAS maintains tumour growth by boosting glucose uptake and transporting glucose intermediates into the pentose phosphate pathway (PPP) and hexosamine biosynthesis pathway (HBP). Nowadays, the interest to understand the reprogramming of metabolism in neoplasic transformation is increasing. Although the molecular mechanism behind the upregulation of glucose metabolism is not yet understood, the pivotal role played by KRAS signalling in the homeostasis of aerobic glycolysis has been reported in several types of cancer. For example, in a PDAC murine model, it has been demonstrated that mutated KRAS keeps tumour growth by stimulating glucose uptake and leading glucose intermediates into the hexosamine biosynthesis pathway (HBP) and pentose phosphate pathway (PPP) (Ying et al. 2012). Remarkably, knockdown of rate-limiting enzymes in HBP or PPP halted tumour growth, indicating their potential as therapeutic targets .
In human colorectal cancer , the increase of glucose transporter 1 (GLUT1) expression and glucose uptake is critically dependent on KRAS mutational state (Yun et al. 2009). In fact, PET (fluorodeoxyglucose (FDG) positron emission tomography scan) is used to evaluate tumour size and location by analysing glucose metabolism by measuring the uptake of FDG, a glucose analogue and it has been described that CRC cells with mutated KRAS show an increased FDG accumulation via of GLUT1 upregulation (Kawada et al. 2012; Iwamoto et al. 2014).
On the other hand, the tight relationship between GLUT-1 and KRAS is confirmed by studies reporting that glucose deprivation contributes to the development of mutated KRAS pathways in tumour cells (Yun et al. 2009).
Actually, GLUT1 has been addressed as a potential target in oncology drug discovery since in 2014 a crystal structure of human GLUT-1 was obtained. This achievement will surely be helpful in the discovery of new GLUT1 inhibitors as anti-cancer agents (Flight 2011). First, GLUT-1 antibodies were shown to inhibit breast cancer and lung cancer (NSCLC) cell lines growth (Rastogi et al. 2007). Interestingly, in 2010 a series of synthetic polyphenolic esters were also described to inhibit glucose transport through the cell membrane, and to exert a certain anti-proliferative activity in the H1299 lung cancer cell line. Moreover, these molecules were used in combination with anti-cancer drugs cisplatin or paclitaxel demonstrating synergistic effects in the inhibition of breast and lung cancer cell growth (Granchi et al. 2016).
Likewise, the small-molecule N-[4-chloro-3-(trifluoromethyl)phenyl]-3-oxobutanamide (fasentin) was identified as a chemical sensitizer to the death receptor stimuli FAS and tumour necrosis factor (TNF) apoptosis-inducing ligand. Fasentin interacts with a unique GLUT-1 site in the intracellular channel of this protein, thus inhibiting glucose transport (Wood et al. 2008)
GLUT-1 also plays an essential role for the homeostasis of pancreatic, ovarian, and glioblastoma cancer stem cells (CSCs). WZB117, a specific GLUT1 inhibitor, was shown to inhibit the self-renewal and tumour-initiating capacity of the CSCs without compromising their proliferative potential in vitro. In vivo, systemic WZB117 administration was able to inhibit tumour initiation after implantation of CSCs without causing significant adverse events in host animals (Shibuya et al. 2015). Recently, WZB117 was shown to kill lung and breast cancer cells by inhibiting GLUT1-mediated glucose transport, leaving non-tumorigenic cells unaffected (Shibuya et al. 2015)
In addition to their glucose dependency, tumour growth and survival also relies on glutamine uptake. Glutamine is a fundamental carbon source for the tricarboxylic acid (TCA) cycle and a nitrogen source for nucleotides and nonessential amino acids. Glutamine is also involved in other cellular processes in cancer cells, such as (mTOR) signalling and including anti-oxidative stress. Therefore, glutamine dependent pathways and signalling involved in cancer cell survival, progression and metastasis is a hot topic in cancer research (Deberardinis and Cheng 2010; Wise and Thompson 2010).
As it has been reviewed for glucose transport, glutamine metabolism exhibits pleiotropic effects on cancer cell signalling and therapeutic suppression of glutamine metabolism is considered to be an attractive anti-cancer strategy.
For instance, Benzylserine and L-γ-glutamyl-p-nitroanilide (GPNA), the inhibitor of the glutamine transporter SLC1A5, have been shown to be effective agents in the treatment of non-small cell lung cancer cell lines and murine xenografts (Hassanein et al. 2015). However, these drugs have been shown to induce unselective toxicity in normal, healthy cells that require glutamine for other pathways.
Some other small inhibitors, such as, CB-839 and bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTE) have been reported to specifically target glutaminase (GLS) isoforms not often expressed in normal cells (Xiang et al. 2015)
In view of the abovementioned factors supporting the tight interplay between mutated KRAS and the Warburg metabolism in cancer and the partial success of molecules directly targeting KRAS, quest for new scopes and molecules capable to overcome chemotherapy resistance in tumours displaying gene mutations downstream EGFR should be considered as a top priority in oncological research worldwide.
3.5 Uncoupling the “Warburg Effect” in CRC: A Chink in the Armour of KRAS Dependent Chemoresistance
As it has been widely explained, CRC is the second leading cause of cancer-related deaths and trials using apoptosis-inducing ligand monotherapy to overcome resistance to apoptosis in colon cancer have not shown clinical benefits.
There is a need for a novel focus to overcome clinical resistance to chemotherapies, mainly due to RAS/RAF mutations.
It has been also explained that the enhanced metabolic requirements of colon cancer cells necessarily involves increased glucose uptake and glycolytic flux relative to normal tissues. This feature is used to visualize colon cancer cells using positron emission tomography (PET) where signals emitted from 2-deoxy-2-fluoro-D-glucose (FDG), which is taken up preferentially by colon cancer cells, are monitored.
Currently, some promising molecules are being investigated to bypass KRAS dependent chemoresistance, using a metabolic approach (Fig. 3.1c).
In 2016, Carr et al., published an interesting article where they overcome colon cancer cells resistance to TRAIL (tumour necrosis factor-related apoptosis-inducing ligand) using 2-deoxy-D-glucose (2DG), which is molecularly similar to FDG and is preferentially uptaken by cancer cells. In tumour cells, 2DG metabolism may affect death receptor (DR) expression (TRAIL is a DR ligand) and dissociate the Bak-Mcl-1 complex in cells with high glycolytic activity (Yamaguchi and Perkins 2012). In addition, 2DG is a receptor-competitive inhibitor of glucose, increasing oxidative stress, inhibiting N-linked glycosylation, and hence inducing autophagy. It inhibits cell growth and facilitates selective apoptosis in cancer cells.
The Ras/Raf/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated (ERK) cascade is involved the signal transduction from the cell surface receptors to the nucleus and regulates cell cycle, cell proliferation, cell differentiation and survival. Genetic and epigenetic alterations in many of the key players in this pathway have been found to be associated with cancer.
The dual RAF/MEK kinase Inhibitor RO5126766, synthesized in Chugai Pharmaceuticals Co., Ltd. was shown to decrease FDG uptake in KRAS and BRAF mutant colon cancer murine xenografts (Tegnebratt et al. 2013). This metabolic inhibition correlates with a decreased cell membrane expression of the glucose receptor GLUT-1 and it was tightly associated with a notably reduced expression of the marker of proliferation Ki67.
In the 70–80th of the twentieth century, several scientific reports demonstrated that the anti-diabetic biguanide drugs phenformin (PF) and buformin (BF) can exert some anti-tumour activity in animal models and increase from 5 to 10-years survival of cancer patients.
Metformin (1,1-dimethylbiguanide hydrochloride) is often used to reduce hepatic gluconeogenesis and increase skeletal muscle glucose uptake in patients with type 2 diabetes. Metformin has been proposed as adjuvant therapy in cancer treatment because of its ability to limit cancer incidence by negatively modulating the PI3K /AKT/mTOR pathway.
In 2015, Jia et al. reported that Metformin might prevent induced colorectal cancer in diabetic rats by reversing the Warburg effect. They described that Warburg inhibition was mediated through inhibition of the master regulator PKM2. In fact, PKM2 metformin-induced inhibition has been also reported in other neoplasia. For instance, Metformin Induces apoptosis, downregulating PKM2 in breast cancer cells grown in poor nutrient conditions (Silvestri et al. 2015).
c-Src, is found to be over-expressed and activated in a wide variety of human tumours. The relationship between Src activation and cancer progression appears to be significant, regulating cancer glucose metabolism in premalignant estrogen receptor (ER)-negative mammary epithelial cells.
In this model, Saracatinib, a highly selective, dual Src/Abl kinase inhibitor, was shown to blocks c-Myc translation and glucose metabolism a result of an inhibition in ERK1/2-MNK1-eIF4E-mediated cap-dependent translation of c-Myc and transcription of the glucose transporter GLUT1, therefore interfering with the Warbug effect.
Saracatinib has been used in a phase II trial treating patients with previously treated metastatic colon cancer or rectal cancer. Results from this study suggested that may stop the growth of tumour cells by blocking blood flow to the tumour and by blocking some of the enzymes needed for cell growth (ClinicalTrials.gov Identifier: NCT00397878).
3.5.1 Vitamin C: Reassessing the Role of Vitamin C in Metabolic Enhanced Colon Cancer
In 1976, Linus Pauling and Ewan Cameron performed a pioneering clinical study of the survival times of 100 terminal cancer patients who were given supplemental ascorbate (usually 10 g/day) plus adjuvant chemotherapy and 1000 matched controls, similar patients who had received the same treatment except for the ascorbate. Survival times greater than 1 year after the date of untreatability were observed for 22% of the ascorbate-treated patients and for 0.4% of the controls (Cameron and Pauling 1976).
Many authors have reported that vitamin C shows certain anti-tumour activity, but in spite of the several efforts carried out in order to unravel the molecular mechanism underlying this killing effect and its intriguing selective activity, this is still far from clear.
Scientific data published by Chen Q et al., supporting the previous clinical study carried out by Pauling and Cameron , have shown that vitamin C exerts killing effects on cancer cells from very different origin, displaying a wide variety of gene mutations (many of them do not display KRAS mutation) and alterations in different signalling pathways (Chen et al. 2008).
In 2015 in an article published in Science, Yun J et al., presented data stating that oxidized vitamin C was able to kill CRC cells depending on the KRAS mutational status (Yun et al. 2015). They found that cultured human CRC cells harbouring KRAS or BRAF mutations were selectively killed when exposed to high levels of vitamin C. This effect was due to increased uptake of the oxidized form of vitamin C, dehydroascorbate (DHA), via the GLUT1 glucose transporter. Increased DHA uptake caused oxidative stress when intracellular DHA is reduced to vitamin C, depleting glutathione. Thus, ROS accumulate and inactivate glyceraldehyde 3-phosphate dehydrogenase (GAPDH), an enzyme of ~37 kDa that catalyses the sixth step of glycolysis and thus serves to break down glucose for energy and carbon molecules. Inhibition of GAPDH in highly glycolytic KRAS or BRAF mutant cells leads to an energetic catastrophe and cell death not seen in normal cells.
Later, in 2016 it was published another scientific work partially confirming previous observations of Yun et al., but mainly focused on the putative interaction of vitamin C in the Warburg metabolism that is considered, as stated previously, a hallmark in cancer and glycolytic enzymes reported as central points of regulation in cancer. In this work we described a novel anti-tumour mechanism of vitamin C in KRAS mutant colorectal cancer involving the Warburg metabolic disruption through downregulation of key metabolic checkpoints in KRAS mutant cancer cells and tumours without killing human immortalized colonocytes (Aguilera et al. 2016). Vitamin C is capable to induce RAS detachment from the cell membrane via ROS inhibition. Thus, RAS detachment leads inhibition ERK 1/2 and PKM2 phosphorylation. As a consequence of this activity, we could observe strong downregulation of the glucose transporter (GLUT-1) and pyruvate kinase M2 (PKM2)-PTB dependent protein expression causing a major blockage of the Warburg effect and therefore energetic stress.
Tumour-specific pyruvate kinase M2 (PKM2) is a master regulator for the Warburg effect and In addition to its well-established role in aerobic glycolysis, PKM2 directly regulates gene transcription [90].
Interestingly, in 2014 Tian et al., published that highly glycolytic cells triggered by activation of the hypoxia-inducible factor (HIF) pathway greatly enhanced vitamin C-induced toxicity in multiple cancer cell lines, including von Hippel-Lindau (VHL)-defective renal cancer cells. According to their observations, HIF increases the intracellular uptake of DHA through its transcriptional target glucose transporter 1 (GLUT1), synergizing with the uptake of its reduced form through sodium-dependent vitamin C transporters (Tian et al. 2014).
The majority of these results point to the Warburg metabolism and related overexpressed enzymes in cancer as a promising scientific field that requires further in-depth studies in order to find new therapeutic targets . Encouraging results observed in vitamin C research, such as its ability to overcome anti-EGFR resistance and displayed selectivity, emphasizes the need for further research on this topic and may open the door to a novel generation of molecules that may constitute a new hope in handling RAS dependent chemoresistant cancer.
3.6 Conclusions
KRAS dependent chemoresistance is a major threat to the clinical handling of CRC and other neoplasia. Among all the CRC subtypes, the CMS3 also called “metabolic subtype”, shows frequent KRAS mutation and concomitant metabolic alterations. Many authors have shown compelling data highlighting the role of KRAS signalling in the regulation of aerobic glycolysis in several types of cancer.
KRAS harbouring cancers have altered metabolism involving enhanced nutrients uptake glycolysis, glutaminolysis, and elevated synthesis of nucleotides and fatty acids.
Unfortunately, clinical trials with molecules targeting KRAS did not render clear benefits to the overall survival (OS) and progression-free survival of metastatic colorectal cancer patients and, to date, no effective treatments that target mutant variants of KRAS have been introduced into clinical practice. It is time to propose different approaches to break down the KRAS barrier to chemotherapy. Counteracting KRAS-ruled metabolic pathways may be a promising focus in order to sensitize KRAS mutant tumours to chemotherapy and many studies are already centred in this approach.
-
But the problem seems to be far more complex.
Glucose requirements and carbon sources in tumours are much more heterogeneous than initially thought. Currently, new studies have been developed showing a dual capacity of tumour cells for glycolytic and oxidative phosphorylation (OXPHOS) metabolism.
Metabolic OXPHOS-dependent cancer cells are capable to use alternative substrates, such as glutamine and/or fatty acids. Therefore, the variety of carbon substrates able to fuel neoplastic cells points out to a high metabolic heterogeneity, even within tumours sharing the stage and clinical diagnosis. Indeed, it has been reported that 80% of the ATP generation in MCF7 breast cancer cells relies on mitochondrial respiration (Guppy et al. 2002).
Furthermore, some studies have reported that glycolysis inhibition often restores OXPHOS in cancer cells (Bonnet et al. 2007; Michelakis et al. 2010) demonstrating that in spite of the augmented glycolytic rates often shown by cancer cells, mitochondrial oxidative metabolism remains intact. The overall data strongly point out to a high cancer metabolic plasticity, implying that molecules targeting metabolic factors in cancer may face similar mechanisms of resistance as previously described for conventional chemotherapy.
Combination of metabolic inhibitors and classical chemotherapeutic agents may constitute a great advance to address KRAS-driven chemoresistance , often attributed to the concomitant altered metabolic expression patterns.
References
Aguilera O, Fraga MF, Ballestar E, Paz MF, Herranz M, Espada J, García JM, Muñoz A, Esteller M, González-Sancho JM (2006) Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene 25:4116–4121. https://doi.org/10.1038/sj.onc.1209439
Aguilera O, Muñoz-Sagastibelza M, Torrejón B, Borrero-Palacios A, Del Puerto-Nevado L, Martínez-Useros J, Rodriguez-Remirez M, Zazo S, García E, Fraga M, Rojo F, García-Foncillas J (2016) Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 7:47954–47965. https://doi.org/10.18632/oncotarget.10087
Akkiprik M, Celikel CA, Düşünceli F, Sönmez O, Güllüoğlu BM, Sav A, Ozer A (2008) Relationship between overexpression of ras p21 oncoprotein and K-ras codon 12 and 13 mutations in Turkish colorectal cancer patients. Turk J Gastroenterol 19:22–27
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11:37–51. https://doi.org/10.1016/j.ccr.2006.10.020
Bray F, Møller B (2006) Predicting the future burden of cancer. Nat Rev Cancer 6:63–74. https://doi.org/10.1038/nrc1781
Cameron E, Pauling L (1976) Supplemental ascorbate in the supportive treatment of cancer: prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A 73:3685–3689
Carr RM, Qiao G, Qin J, Jayaraman S, Prabhakar BS, Maker AV (2016) Targeting the metabolic pathway of human colon cancer overcomes resistance to TRAIL-induced apoptosis. Cell Death Discov 12:16067. https://doi.org/10.1038/cddiscovery.2016.67
Cejas P, López-Gómez M, Aguayo C, Madero R, de Castro Carpeño J, Belda-Iniesta C, Barriuso J, Moreno García V, Larrauri J, López R, Casado E, Gonzalez-Barón M, Feliu J (2009) KRAS mutations in primary colorectal cancer tumors and related metastases: a potential role in prediction of lung metastasis. PLoS One 4:e8199. https://doi.org/10.1371/journal.pone.0008199
Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, Khosh DB, Drisko J, Levine M (2008) Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A 105:11105–11109. https://doi.org/10.1073/pnas.0804226105
Conlin A, Smith G, Carey FA, Wolf CR, Steele RJ (2005) The prognostic significance of K-ras, p53, and APC mutations in colorectal carcinoma. Gut 54:1283–1286. https://doi.org/10.1136/gut.2005.066514
Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ (2014) Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 13:828–851. https://doi.org/10.1038/nrd4389
Dang CV (2012) Links between metabolism and cancer. Genes Dev 26:877–890. https://doi.org/10.1101/gad.189365
Dang CV, Kim JW, Gao P, Yustein J (2008) The interplay between MYC and HIF in cancer. Nat Rev Cancer 8:51–56. https://doi.org/10.1038/nrc2274
Dayton TL, Jacks T, Vander Heiden MG (2016) PKM2, cancer metabolism, and the road ahead. EMBO Rep 17:1721–1730. https://doi.org/10.15252/embr.201643300
De Berardinis RJ, Cheng T (2010) Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29:313–324. https://doi.org/10.1038/onc.2009.358
Ferlay J, Soerjomataram I, Dikshit R, Eser S Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 5:359–386. https://doi.org/10.1002/ijc.29210
Flight MH (2011) A sweet blow for cancer cells. Nat Rev Drug Discov 10:734. https://doi.org/10.1038/nrd3566
Granchi C, Fortunato S, Minutolo F (2016) Anticancer agents interacting with membrane glucose transporters. Med Chem Comm 7:1716–1729. https://doi.org/10.1039/C6MD00287K
Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa E, Melo F, Missiaglia E, Ramay H, Barras D, Homicsko K, Maru D, Manyam GC, Broom B, Boige V, Perez-Villamil B, Laderas T, Salazar R, Gray JW, Hanahan D, Tabernero J, Bernards R, Friend SH, Laurent-Puig P, Medema JP, Sadanandam A, Wessels L, Delorenzi M, Kopetz S, Vermeulen L, Tejpar S (2015) The consensus molecular subtypes of colorectal cancer. Nat Med 11:1350–1356. https://doi.org/10.1038/nm.3967
Guppy M, Leedman P, Zu X, Russell V (2002) Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J 364:309–315. https://doi.org/10.1042/bj3640309
Hassanein M, Qian J, Hoeksema MD, Wang J, Jacobovitz M, Ji X, Harris FT, Harris BK, Boyd KL, Chen H, Eisenberg R, Massion PP (2015) Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int J Cancer 137:158715–158797. https://doi.org/10.1002/ijc.29535
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350:2335–2342. https://doi.org/10.1056/NEJMoa032691
Irahara N, Baba Y, Nosho K, Shima K, Yan L, Dias-Santagata D, Iafrate AJ, Fuchs CS, Haigis KM, Ogino S (2010) NRAS mutations are rare in colorectal cancer. Diagn Mol Pathol 19:157–163. https://doi.org/10.1097/PDM.0b013e3181c93fd1
Iwamoto M, Kawada K, Nakamoto Y, Itatani Y, Inamoto S, Toda K, Kimura H, Sasazuki T, Shirasawa S, Okuyama H, Inoue M, Hasegawa S, Togashi K, Sakai Y (2014) Regulation of 18F-FDG accumulation in colorectal cancer cells with mutated KRAS. J Nucl Med 55:2038–2044. https://doi.org/10.2967/jnumed.114.142927
Jia Y, Ma Z, Liu X, Zhou W, He S, Xu X, Ren G, Xu G, Tian K (2015) Metformin prevents DMH-induced colorectal cancer in diabetic rats by reversing the Warburg effect. Cancer Med 4:1730–1741. https://doi.org/10.1002/cam4.521
Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ (2007) Cetuximab for the treatment of colorectal cancer. N Engl J Med 357:2040–2048. https://doi.org/10.1056/NEJMoa071834
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359:1757–1765. https://doi.org/10.1056/NEJMoa0804385
Kawada K, Nakamoto Y, Kawada M, Hida K, Matsumoto T, Murakami T, Hasegawa S, Togashi K, Sakai Y (2012) Relationship between 18F-fluorodeoxyglucose accumulation and KRAS/BRAF mutations in colorectal cancer. Clin Cancer Res 18:1696–1703. https://doi.org/10.1158/1078-0432.CCR-11-1909
Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti LMP (2017) Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14:11–31. https://doi.org/10.1038/nrclinonc.2017.1
Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:m31ra34. https://doi.org/10.1126/scitranslmed.3000677
Morani F, Phadngam S, Follo C, Titone R, Aimaretti G, Galetto A, Alabiso O, Isidoro C (2014) PTEN regulates plasma membrane expression of glucose transporter 1 and glucose uptake in thyroid cancer cells. J Mol Endocrinol 53:247–258. https://doi.org/10.1530/JME-14-0118
Nash GM, Gimbel M, Cohen AM, Zeng ZS, Ndubuisi MI, Nathanson DR, Ott J, Barany F, Paty PB (2010) KRAS mutation and microsatellite instability: two genetic markers of early tumor development that influence the prognosis of colorectal cancer. Ann Surg Oncol 17:416–424. https://doi.org/10.1245/s10434-009-0713-0
Prior IA, Lewis PD, Mattos C (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res 72:2457–2467. https://doi.org/10.1158/0008-5472.CAN-11-2612
Rastogi S, Banerjee S, Chellappan S, Simon GR (2007) Glut-1 antibodies induce growth arrest and apoptosis in human cancer cell lines. Cancer Lett 257:244–251. https://doi.org/10.1016/j.canlet.2007.07.021
Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, Kitanaka C (2015) Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 6:651–661. https://doi.org/10.18632/oncotarget.2892
Shimizu N, Ohtsubo M, Minoshima S (2007) Mutation view/KM cancer DB: a database for cancer gene mutation. Cancer Sci 98:259–267. https://doi.org/10.1111/j.1349-7006.2007.00405
Silvestri A, Palumbo F, Rasi I, Posca D, Pavlidou T, Paoluzi S, Castagnoli L, Cesareni G (2015) Metformin induces apoptosis and downregulates pyruvate kinase M2 in breast cancer cells only when grown in nutrient-poor conditions. PLoS One 10(8):e0136250. https://doi.org/10.1371/journal.pone.0136250
Tegnebratt T, Lu L, Lee L, Meresse V, Tessier J, Ishii N, Harada N, Pisa P, Stone-Elander S (2013) [18 F]FDG-PET imaging is an early non-invasive pharmacodynamic biomarker for a first-in-class dual MEK/Raf inhibitor, RO5126766 (CH5126766), in preclinical xenograft models. EJNMMI Res 3(1):67. https://doi.org/10.1186/2191-219X-3-67
Tian W, Wang Y, Xu Y, Guo X, Wang B, Sun L, Liu L, Cui F, Zhuang Q, Bao X, Schley G, Chung TL, Laslett AL, Willam C, Qin B, Maxwell PH, Esteban MA (2014) The hypoxia-inducible factor renders cancer cells more sensitive to vitamin C-induced toxicity. J Biol Chem 289:3339–3351. https://doi.org/10.1074/jbc.M113.538157
Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360:1408–1417. https://doi.org/10.1056/NEJMoa0805019
Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS (2011) Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer 50:307–312. https://doi.org/10.1002/gcc.20854
Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL (1988) Genetic alterations during colorectal-tumor development. N Engl J Med 319:525–532. https://doi.org/10.1056/NEJM198809013190901
Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, Zhang Y, You MJ, Koochekpour S, Saleem M, Huang H, Lu J, Deng Y (2014) Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep 8:1461–1474. https://doi.org/10.1016/j.celrep.2014.07.053
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21:297–308. https://doi.org/10.1016/j.ccr.2012.02.014
Wise DR, Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35:427–433. https://doi.org/10.1016/j.tibs.2010.05.003
Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, Gronda M, Eberhard Y, Minden MD, Bilan PJ, Klip A, Batey RA, Schimmer AD (2008) A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol Cancer Ther 7:3546–3555. https://doi.org/10.1158/1535-7163.MCT-08-0569
Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, Hsieh AL, Gouw AM, Thomas AG, Gao P, Sun L, Song L, Yan B, Slusher BS, Zhuo J, Ooi LL, Lee CG, Mancuso A, McCallion AS, Le A, Milone MC, Rayport S, Felsher DW, Dang CV (2015) Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest 125:2293–2306. https://doi.org/10.1172/JCI75836
Yamaguchi R, Perkins G (2012) Finding a panacea among combination cancer therapies. Cancer Res 72:18–23. https://doi.org/10.1158/0008-5472.CAN-11-3091
Ye LC, Liu TS, Ren L, Wei Y, Zhu DX, Zai SY, Ye QH, Yu Y, Xu B, Qin XY, Xu J (2013) Randomized controlled trial of cetuximab plus chemotherapy for patients with KRAS wild-type unresectable colorectal liver-limited metastases. J Clin Oncol 31:1931–1938. https://doi.org/10.1200/JCO.2012.44.8308
Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149:656–670. https://doi.org/10.1016/j.cell.2012.01.058
Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C, Stephens P, Edkins S, Tsui WW, Chan AS, Futreal PA, Stratton MR, Wooster R, Leung SY (2002) Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res 62:6451–6455
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagolapan H, Schmidt K, Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N (2009) Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325(5947):1555–1559. https://doi.org/10.1126/science.1174229
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N (2015) Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325:1555–1559. https://doi.org/10.1126/science.1174229
Zavodna K, Konecny M, Krivulcik T, Spanik S, Behulova R, Vizvaryova M, Weismanova E, Galbavy S, Kausitz J (2009) Genetic analysis of KRAS mutation status in metastatic colorectal cancer patients. Neoplasma 56(3):275–278. https://doi.org/10.4149/neo_2009_03_275
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Aguilera, O., Serna-Blasco, R. (2018). Targeting KRAS Mutant CMS3 Subtype by Metabolic Inhibitors. In: Jordan, P. (eds) Targeted Therapy of Colorectal Cancer Subtypes. Advances in Experimental Medicine and Biology, vol 1110. Springer, Cham. https://doi.org/10.1007/978-3-030-02771-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-02771-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-02770-4
Online ISBN: 978-3-030-02771-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)