
Abstract
In 1949, Naffziger et al. first described idiopathic intracranial hypertrophic pachymeningitis (IIHP) as an aseptic, diffuse inflammatory disease that causes thickening of the dura mater and often headache and progressive multiple nerve palsies due to fibrous entrapment or ischemic damage of neurovascular structures. Pachymeningeal thickening can be diffuse or nodular. We report two cases of IIHP; one was affected by diffuse IIHP, while the other presented focal IIHP mimicking a convexity meningioma. We examine the differential diagnosis between IIHP and other known causes of hypertrophic pachymeningitis. We also discuss the clinical bases of treatment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In 1949, Naffziger et al. first described idiopathic intracranial hypertrophic pachymeningitis (IIHP) as an aseptic, diffuse inflammatory disease causing thickening of the dura mater and often headache and progressive multiple nerve palsies due to fibrous entrapment or ischemic damage of neurovascular structures [1]. It is a rare pathology, and the diagnosis is made by excluding other known causes of hypertrophic pachymeningitis [2, 3, 4, 5].
Idiopathic intracranial hypertrophic pachymeningitis can be diffuse to the whole intracranial pachymeninx or affect the dura mater focally. Focal IIHP more often affects the dura mater of tentorium, falx [6, 7, 8, 9, 10, 11, 12, 13], and parasellar and cavernous sinus [8, 13, 14, 15, 16], clinically and radiologically mimicking a skull-base en plaque meningioma. An IIHP of the convexity is rare, radiologically resembling meningioma.
We report two cases of IIHP, the first diffuse and the second focal and mimicking a convexity meningioma. We examine the differential diagnosis between IIHP and other known causes of hypertrophic pachymeningitis. Further, we discuss the clinical bases of treatment.
Case 1
The patient B.G. had had a history of many hospital admissions for autoimmune pathologies. She was admitted in 1967 for ulcerative colitis and in 1977 for erythema nodosum and polyarthritis with migrant pains. She showed right ear perichondritis in 1983 and right eye iridocyclitis in 1989. During an acute phase of ulcerative colitis, human leukocyte antigen (HLA) typing (a26–a30, b13–b49, cw6-cw7) was done. Autoimmune thyroiditis with antimicrosomial positivity (161.5 UI/ml, normal 0–100) and antiperoxidase antibodies (137.0) were found. Serum values for antinuclear antibodies (ANA) and rheumatoid factor were negative.
Between 1994 and 1999, the patient had multiple admittances to our neurology department for headache, relapsing cranial nerve palsies (III, IV, IX, X, and XII), and left optic neuritis. Neuroradiologic (CT and MRI) and CSF exams were negative. During the hospital stays, erythrocyte sedimentation rate (ESR) (64–100), immunoglobulin M (393 mg%, normal 56–352), and C4 (41.3 mg/dl, normal 12–36) were found to be elevated. Positivity of antimicrosomial and antiperoxidase antibodies was also confirmed. According to the negative neuroradiologic exams and the systemic autoimmune laboratory pattern, multiple cranial nerve palsies were treated by corticosteroid therapy, bringing a transient improvement.
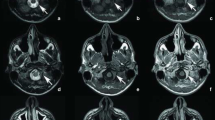
The patient was admitted again to our neurosurgical department because of jacksonian convulsion in the left hemiface and arm. We found a right parietal lesion that was iso- and hypointense on T1-weighted MRI and hypointense on T2-weighted imaging, with marked enhancement after gadolinium (Figs. 1, 2, 3, 4).

Sagittal MRI showing nodular pachymeningitis in case 1

Sagittal MRI showing nodular pachymeningitis in case 1 after administration of gadolinium

Axial MRI of case 1

Axial MRI showing case 1 after administration of gadolinium
During removal of the lesion, the dura mater appeared yellowish and about 2 cm thick within a 4×4-cm area. The dural thickening extended to the superior longitudinal sinus, involving a wide rolandic vein. Dissecting it from the arachnoid plane was easy, and the lesion was completely removed. Histologic examination showed pachymeningeal fragments with an infiltrate of neutrophils, lymphocytes, and plasma cells and delimited by a fibroblastic reaction. Histologic findings matched the diagnosis of pachymeningitis.
Suspecting an autoimmune disease, we checked the serum for Borrelia burgdorferi, Listeria monocytogenes, common bacteria, and fungi. We evaluated Tyne’s test, direct examination, culture of sputum, Mantoux reaction, and the serum value of angiotensin-converting enzyme. All of these exams were negative. We made the diagnosis of IIHP with systemic autoimmune hyperactivity. The postoperative course was uneventful and the patient was rapidly discharged.
Case 2
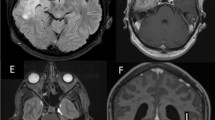
The patient V.A., a 70-year-old female, was admitted to our observation because of a 1-year headache and urinary incontinence. Magnetic resonance imaging showed diffuse pachymeningeal thickening, mainly supratentorial, with intense enhancement after gadolinium administration (Figs. 5, 6). Serologic parameters revealed little alteration. The ESR value was 18 mm/h, and white blood cells and neutrophils were slightly elevated. A CSF culture and biopsy were negative. Cerebrospinal fluid examination showed elevated protein (112 mg/dl) and glucose (133 mg/dl).

Sagittal MRI showing diffuse pachymeningitis in case 2

Sagittal MRI of case 2 after administration of gadolinium, showing diffuse pachymeningitis
The patient underwent right frontal craniotomy with biopsy of the dura mater, which appeared moderately thickened. Histologic examination showed thickened pachymeningeal fragments, with blood vessels and local hemorrhagic deposit surrounded by siderophages. The histologic diagnosis was of IIHP (Fig. 7).

Pachymeningeal fragments, with an infiltrate of neutrophils, lymphocytes, and plasma cells, delimited by a fibroblastic reaction
Discussion
Idiopathic intracranial hypertrophic pachymeningitis is a chronic inflammatory disease that causes thickening of the dura mater that can involve cranial bones, pericranial soft tissue [13, 17, 18, 19] and, rarely, the cerebral parenchyma, infiltrated through the Virchow-Robin spaces by the inflammatory cells [15, 18].
Pachymeningeal thickening can be diffuse or nodular. Deprez et al. [3] and Hatano et al. [1] describe two main groups of nodular hypertrophic pachymeningitis according to the region involved and the pattern of dural thickening. The first group includes cases with involvement from the parasellar and cavernous sinus to the superior orbital fissure, while the second group shows falcotentorial and clival involvement. Idiopathic intracranial hypertrophic pachymeningitis of the cerebral convexity occurs much more rarely. Among 73 reported cases, only 11 [1, 3, 8, 18, 19, 20], including our case 1, presented as convexity pseudotumoral dural thickening.
Histologically, dural thickening shows a nonspecific inflammatory reaction in multiple phases of evolution, with a fibroblastic infiltrate of neutrophils, lymphocytes, and plasma cells [3, 8, 9, 15, 17, 19]. The diagnosis of IIHP is made by exclusion, since many pathologies can cause acute or chronic inflammatory thickening of the dura mater. Therefore, extensive diagnostic procedures, including dural biopsy, are mandatory to exclude systemic pathologies.
Neurosarcoidosis can cause dural thickening [3, 4, 5, 13, 15, 18, 19] and involves the central nervous system in 5% of cases [21, 22, 23]. In both cases reported here, the normal values of serum angiotensin-converting enzyme and the absence of noncaseous granulomas excluded this diagnosis.
Mycobacterium tuberculosis can be a cause of hypertrophic pachymeningitis [3, 5, 13, 18, 24]; in both our cases, no tuberculomas with Langhans’ cells were found, and Ziehl-Neelsen stain and Löwenstein-Jenson culture were negative.
Hypertrophic pachymeningitis can also be due to fungal infection [25] (histopathologic features and CSF exam excluded this diagnosis in our cases) or neurosyphilis [10, 14, 21, 22, 23, 26, 27], which we excluded this because of negative serologic tests and the absence of histopathologically specific vasculitis. We excluded pachymeningitis due to rheumatoid arthritis because of the absence of articular symptoms. Serum values were negative for ANA and rheumatoid factor. Wegener’s granulomatosis was excluded since the chest radiograph was normal and because of the absence of necrotizing vasculitis on histopathologic examination.
Histopathologic examination also excluded malignant tumors or benign lesions as meningioma en plaque. Magnetic resonance imaging examination allowed us to exclude spontaneous intracranial hypotension. Case 1 presented nodular pachymeningeal thickening, and case 2 showed diffuse thickening of the pachymeninx, but there was no caudal shift of the brainstem or subdural fluid retention. The clinical symptoms were not related to intracranial hypotension.
The mean reported age is 50 years and ranges between 20 and 80 [5], with peak age in the sixth decade [2]. The M:F ratio is about 1.2:1 [5]. The clinical presentation is nonspecific and variable; headache, cranial nerve palsies, and ataxia are the most common symptoms [3, 14, 15, 17, 19]. Nodular IIHP from the parasellar and cavernous sinus to the superior orbital fissure frequently shows supraclinoidal carotid and II, III, IV, V, and VI cranial nerve involvement. Falcotentorial and clival IIHP can involve the cranial nerves V, VII, VIII, IX, and X [1]. Rarely, IIHP of the posterior cranial fossa has been reported to lead to internal carotid artery occlusion [13, 16] or hydrocephalus [3, 9, 15, 17, 24, 28, 29]. Nishizaki et al. [18] described a symptomatologic triad as in our case 1, presenting with headache, multiple cranial nerve palsies, and seizure.
Multiple cranial nerve neuropathies are due to their compression at skull-base foramina by thickened pachymeninges [13]. Compressive carotid occlusion by extrinsic stenosis at the skull base can lead to cortical deficits [16]. Such lesions have a mass effect, but inflammatory perivascular infiltration also plays an important role in cortical irritative symptomatology [15, 18].
Case 1 showed pachymeningeal thickening involving the rolandic vein and extending to the superior sagittal sinus. The infiltration or complete occlusion of the dural sinuses has been frequently reported [4, 13, 30, 31]. Hamada et al. [24] reported a case of dural arteriovenous fistula attributable to falcotentorial IIHP; they maintained that the arteriovenous fistula was due to chronic stenosis and hypertension of the straight sinus and dural tentorial veins by venous inflammatory infiltration. Blood count and serum chemistries showed increased ESR in nearly all cases reported [4, 5] and elevated protein levels in about 60%; CSF examination showed pleocytosis in about 25% [4, 5].
Magnetic resonance imaging exam is characteristic and shows various degrees of the lesion’s inflammatory pattern correlating with clinical outcome and prognosis [4, 5]. T1-weighted images show iso- or hypo-intense thickened dura [1, 2, 4, 18, 19] with intense enhancement [1, 32]. The dura mater appears hypointense in T2-weighted images, surrounded by the thin hyperintense border of the lesion [1, 4, 8, 17, 18, 33]; the central hypointense area is attributed to the fibrous reacting tissue and the peripheral border to the acute inflammatory process [1, 18].
Relating to clinical course and prognosis, MRI can show two patterns. The first, with diffuse and homogeneous dural thickening and well-defined T2-weighted borders, frequently correlates to short clinical course and best prognosis. The second pattern, related to cases with chronic course and unfavorable prognosis, shows a hypointense hypertrophic meninx on T2-weighted scans, without a hyperintense border and with dishomogeneous enhancement probably due to cellular compactness and the abundant collagenous materials [4].
In diffuse or skull-base localized IIHP, clinical/instrumental diagnosis is possible (positive MRI and effectiveness of corticosteroid therapy), while in rare nodular IIHP with a convexity site and doubts about diagnosis, biopsy is essential [4, 13] to exclude other neoplastic lesions and en plaque meningioma. Otherwise, surgical approach to nodular IIHP of the convexity permits total removal of the lesion.
Corticosteroid therapy should be the first approach in diffuse or skull-base localized IIHP [1, 4, 18]. Some authors [3, 8, 18] describe only a partial effectiveness of corticosteroid therapy, with transient clinical improvement and relapse when tapering the dosage, followed by steroid-dependent onset. To avoid corticosteroid dependence and side effects associated with long-term therapy, some authors [1, 4] propose steroid pulse therapy with methylprednisolone (1000 mg/day for 3 days). Other authors, considering an autoimmune pathogenesis, administered immunomodulators such as azathioprine and cyclophosphamide [1, 3, 8, 20, 33].
Parasellar IIHP with optic nerve compression and rapid visual loss can justify emergent surgical excision. However, such cases are rare [13, 14, 24, 34], and some authors [24] think it is due to optic nerves’ compression and also their infiltration by inflammatory tissue. Corticosteroid therapy is ineffective [24], and surgical decompression is required.
Parney et al. [5] considered a case of hypertrophic pachymeningitis to be a pattern of occult tuberculosis that was successfully treated with antituberculous therapy. However, empirical antituberculous treatment has been unsuccessful in other cases [1]. Considering nodular IIHP of the convexity as a rare pathology, the clinicoradiologic suspicion of a convexity meningioma associated with systemic hyperactivity was correct in our case 1. As Deprez et al. [3] reported, too, there is a clear association between IIHP and a disease not fitting the criteria of specific autoimmune pathology but clearly indicating a dysimmune status [35]. This pattern is the etiopathogenetic basis of the lesion we found. In fact, this disease probably represents the result of an autoimmune process.
In clinical presentation, ANA positivity and elevated ESR are frequent [3, 36]. As in our case 1, the association with well-defined autoimmune diseases such as rheumatoid arthritis [37, 38, 39, 40], polyarteritis nodosa [40], and Wegener’s granulomatosis [41, 42] is often reported [3, 43]. Furthermore, the HLA typing is often analogous to that of patients affected by autoimmune disease [3].
The natural history of IIHP is not well known [13], and spontaneous resolution has been reported [44]. However, among the cases successfully treated with corticosteroid therapy, the prognosis is favorable, with radiologic or clinical resolution of the disease. These cases show MRI with well-defined hyperintense borders on T2-weighted scan and homogeneous enhancement on T1-weighted images.
Steroid drug dependence plays an important role for the prognosis in case of relapse: if the patient experiences relapse with corticosteroid independence, the prognosis is still good. Patients improve again with administration of corticosteroid therapy, and the pathology can become chronic when therapy is tapered [1]. If relapse occurs with corticosteroid dependence, the prognosis is less favorable. High doses of corticosteroid can lead the pathology to an inactive phase and to become corticosteroid-independent [1]. In such cases, during active phases of the disease, therapy must be directed to preventing irreversible cranial nerve neuropathies [1].
References
Hatano N, Behari S, Nagatani T, Kimura M, Ooka K, Saito K, Yoshida J (1999) Idiopathic hypertrophic cranial pachymeningitis: clinicoradiological spectrum and therapeutic options. Neurosurgery 45:1336–1344
Nishioka H, Ito H, Haraoka J, Yamada Y, Nojima H (1998) Idiopathic hypertrophic cranial pachymeningitis with accumulation of Tallium-201 on single photon emission CT. Am J Neuroradiol 19:450–453
Deprez M, Born J, Hauwaert C, Otto B (1997) Idiopathic hypertrophic cranial pachymeningitis mimicking multiple meningiomas: case report and review of the literature. Acta Neuropathol 94:385–389
Bang OY, Kim DI, Yoon SR, Choi IS (1998) Idiopathic hypertrophic pachymeningeal lesions. Eur Neurol 39:49–56
Parney IF, Johnson ES, Allen PBR (1997) Idiopathic cranial hypertrophic pachymeningitis responsive to antituberculous therapy: case report. Neurosurgery 41:965–971
Berger JR, Snodgras S, Glaser J, Post MJD, Noremberg M, Benedetto P (1989) Multifocal fibrosclerosis with hypertrophic intracranial pachymeningitis. Neurology 39:1345–1349
Kobayashi N, Hongo K, Kawauchi M, Kobayashi S, Sugita K (1985) Chronic meningitis with marked unilateral tentorial pachymeningitis. Surg Neurol 23:529–535
Mamelak AN, Kelly WM, Davis RL (1993) Idiopathic hypertrophic cranial pachymeningitis. Report of three cases. J Neurosurg 79:270–276
Masson C, Henin D, Decroix JP, Martin N, Cambier J, Masson M (1989) Pachyméningites craniennes de cause indéterminée: étude de trois cas. Rev Neurol (Paris) 145:16–23
Michel D, Girard PF, Tommasi M, Masson R, Trillet M, Piccinali JP (1969) Les pachyméningites granulomateuses intra-craniennes à symptomatologie pseudotumorale. A propos de 4 observations. J Med Lyon 50:547–577
Murai H, Kira J, Kobayashi T, Goto I, Inoue H, Hasuo K (1992) Hypertrophic cranial pachymeningitis due to Aspergillus flavus. Clin Neurol Neurosurg 94:247–250
Naffziger HC, Stern WE (1949) Chronic pachymeningitis: report of a case and review of the literature. Arch Neurol Psychiatry 62:383–404
Goyal M, Lalik A, Mishara NK, Gaikwad SB (1997) Idiopathic hypertrophic pachymeningitis: spectrum of the disease. Neuroradiology 39:619–623
Feringa ER, Weatherbee L (1975) Hypertrophic granulomatous cranial pachymeningitis causing progressive blindness in a chronic dialysis patient. J Neurol Neurosurg Psychiatry 38:1170–1176
Kadoya C, Soejima T, Yamada H, Yokota A (1993) Pachymeningoencephalitis: case report. Neurosurgery 33:131–135
Willing SJ, Broghamer W (1992) Internal carotid artery occlusion due to idiopathic cranial pachymeningitis. AJNR 13:1594, 1596
Botella C, Orozco M, Navarro J, Riesgo P (1994) Idiopathic chronic hypertrophic craniocervical pachymeningitis: case report. Neurosurgery 35:1144–1149
Nishizaki T, Iwamoto F, Uesugi S, Akimura T, Yamashita K, Ito H (1997) Idiopathic cranial pachymeningoencephalitis focally affecting the parietal dura mater and adjacent brain parenchyma: case report. Neurosurgery 40:840–843
Kioumehr F, Au A, Rooholamini SA, Yaghmai I, Verma R (1994) Idiopathic hypertrophic cranial pachymeningitis: a case report. Neuroradiology 36:292–294
Masson C, Henin D, Hauw JJ, Rey A, Raverdy P, Masson M (1993) Cranial pachymeningitis of unknown origin: a study of seven cases. Neurology 43:1329–1334
Cahill DW, Salemon M (1981) Neurosarcoidosis: a review of the rare manifestations. Surg Neurol 15:204–211
De Tribolet N, Zander E, Phil D (1978) Intracranial sarcoidosis presenting angiographically as a sub-dural hematoma. Surg Neurol 9:169–171
Hayes WS, Sherman JL, Stern BJ, Citrin CM, Pulaski PD (1987) MR and CT evaluation of intracranial sarcoidosis. AJNR 8:841–847
Hamada J, Yoshinaga Y, Korogi Y, Ushio Y (2000) Idiopathic hypertrophic cranial pachymeningitis associated with a dural arteriovenous fistula involving the straight sinus: case report. Neurosurgery 47:1230–1233
Gorrel JM, Palutke WA, Chason JL (1979) Candica pachymeningitis with multiple cranial nerve paresis. Arch Neurol 36:719–720
Tyrrel RL II, Brundschuh CV, Modic MT (1987) Dural carcinomatosis: MR demonstration. J Comput Assist Tomogr 11:329–332
Moore AP, Rolfe EB, Jones EL (1985) Pachymeningitis cranialis hypertrophica. J Neurol Neurosurg Psychiatry 48:942–944
Oku T, Yamashita M, Inoue T (1995) A case of posterior fossa hypertrophic pachymeningitis with hydrocephalus. No To Shinkei 47:569–573
Nohra G, Maarrawi J, Samaha E, Rizk T, Okais N (2000) Craniospinal pachymeningitis. Acta Neurochir (Wien) 142:713–714
Takahashi M, Sasajima T, Mineura K, Itoh Y, Kowada M, Iwaya K, Hatazawa J, Ogawa T, Okudera T, Murakami M, Uemura K (1996) Positron emission tomographic evaluation for frontal hypertrophic cranial pachymeningitis using 11C-methyl-L-methionine. No Shinkei Geka 24:287–293
Yunten N, Oran I, Calli C, Parildar M (1999) Hypertrophic cranial pachymeningitis involving dural sinuses: a pseudo signal-void appearance on MRI. Eur J Radiol 31:188–92
Lam BL, Barret DA, Glaser JS, Schatz NJ, Brown HH (1994) Visual loss from idiopathic intracranial pachymeningitis. Neurology 44:694–698
Martin N, Masson C, Henin D, Dompoint D, Marsault C, Nahum H (1989) Hypertrophic cranial pachymeningitis: assessment with CT and MR imaging. AJNR 3:477–484
Harada T, Ohashi T, Ohki K (1996) Optic neuropathy associated with hypertrophic cranial pachymeningitis. Br J Ophthalmol 80:574–575
Fujimoto M, Kira J, Murai H, Yoshimura T, Takizawa K, Goto I (1993) Hypertrophic cranial pachymeningitis associated with mixed collagen tissue disease; a comparison study with idiopathic and infectious pachymeningitis. Intern Med 32:510–512
Tanaka M, Suda M, Ishikawa Y, Fujitake J, Fujii H, Tatsuoka Y (1996) Idiopathic hypertrophic cranial pachymeningitis associated with hydrocephalus and myocarditis: remarkable steroid-induced remission of hypertrophic dura mater. Neurology 46:554–556
Spurlock RG, Richman AV (1983) Rheumatoid meningitis. Arch Pathol Lab Med 107:129–131
Weinstein GW, Powell SR, Thrush WP (1987) Chiasmal neuropathy secondary to rheumatoid pachymeningitis. Am J Ophthalmol 104:439–440
West GS, Pittman DL, Coggin JT (1980) Intracranial plasma cell granuloma. Cancer 46:330–335
Astrom KE, Lidholm SO (1963) Extensive intracranial lesions in a case of orbital non-specific granuloma combined with polyarteritis nodosa. J Clin Pathol 16:137–143
Ghilain S, Delreux V, Kevers L, Sindic CJM, Mathurin P, Laterre EC (1988) Atteinte multiple des paires craniennes associée à une pachymeningite tentorielle à charactère granulomateux. Acta Neurol Belg 88:91–100
Nishino H, Rubino FA, Parisi JE (1993) The spectrum of neurology involvement in Wegener’s granulomatosis. Neurology 43:1334–1337
Agdal N, Hagdrup HK, Wantzin GL (1980) Pachymeningitis cervicalis hypertrophica syphilitica. Acta Derm Venereol 60:184–186
Nishio S, Morioka T, Togawa A, Yanase T, Nawata H, Fukui M, Hasuo K (1995) Spontaneous resolution of hypertrophic cranial pachymeningitis. Neurosurg Rev 18:201–204
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
D’Andrea, G., Trillò, G., Celli, P. et al. Idiopathic intracranial hypertrophic pachymeningitis: two case reports and review of the literature. Neurosurg Rev 27, 199–204 (2004). https://doi.org/10.1007/s10143-004-0321-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10143-004-0321-1