
Abstract
Environment-sensitive genic male sterility is a valid tool for hybrid production and hybrid breeding, but there are no previous reports of the molecular mechanism of fertility conversion. In this study, RNA-seq, phenotypic and cytological observations, and physiological indexes were applied to analyze thermo-sensitive genic male sterility line 4110S under different temperature conditions to explore the fertility transformation mechanism. In total, 3420 differentially expressed genes (DEGs) were identified comprising 2331 upregulated genes and 1089 downregulated genes. The DEGs were apparently distributed among 54 Gene Ontology functional groups. The phenylpropanoid, long-chain fatty acid, and jasmonic acid (JA) biosynthesis pathways were related to male sterility, where their downregulation blocked the synthesis of sporopollenin and JA. Phenotypic and cytological analyses showed that pollen wall defects and anther indehiscence at high temperatures induced sterility. Moreover, enzyme-linked immunosorbent assay results indicated that the abundance of JA was lower in 4110S under restrictive conditions (high temperature) than permissive conditions (low temperature). A possible regulated network of pathways associated with male sterility was suggested. These results provided insights into the molecular mechanism of fertility conversion in the thermosensitive male sterility system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hybrid vigor or heterosis is a natural phenomenon where the progeny derived from interspecific crossings or the same species with different genetic backgrounds outperform their parents in terms of many characteristics such as production and adaptability (Meyer et al. 2004; Fu et al. 2015). Heterosis has been applied widely in some major crops such as maize (Chen and Pan 2001), rice (Deng and He 2017), rapeseed (Zhang 2000), and sunflower (Liu et al. 2006). The utilization of heterosis in crops contributes significantly to the global food supply. Wheat (Triticum aestivum L.) is a vital grain crop and heterosis has been utilized to improve wheat production in some cases (Geng et al. 2018). In particular, the two-line hybrid wheat system based on environment-sensitive genic male sterility (EGMS) is the most efficient and simplest to implement, where it is based mainly on certain light and temperature conditions (Yang et al. 2018). For example, according to the specific period of daylight or the environmental temperature, photo-thermosensitive male sterile plants can self-fertilize or be employed to obtain hybrid seeds as female parents (Xu and Kang 2001). These EGMS lines are crucial for the utilization of heterosis in wheat because of their advantages in terms of an uncomplicated hereditary basis, excellent sterility and restoration properties, and capacity to yield good crossing combinations (Fu et al. 2010). The mechanism of EGMS related to pollen abortion has been studied using physiological (Ju et al. 2018), cytological (Meng et al. 2016), and molecular biology techniques (Jiang and Shi 2009). The rapid development of high-throughput transcriptome sequencing means that wheat has been analyzed at the transcription level to determine the characteristics of pollen abortion and the gene expression profiles associated with male sterility. This technique has been applied widely to many different types of male abortion, such as genic male sterility (Jeong et al. 2014; Chen et al. 2015), cytoplasmic male sterility (An et al. 2014; Li et al. 2015; Ye et al. 2017), and EGMS (Tang et al. 2012).
Male sterility is mainly caused by pollen wall defects, anther dehiscence, and other mechanisms. Successful pollen wall formation is a necessary prerequisite for the normal development of pollen grains. The main pollen wall component is sporopollenin, which is a dimer formed via the polymerization of phenolics and long-chain aliphatic acids (Dobritsa et al. 2009). These two types of substances are generated by the phenylpropanoid and long-chain fatty acid biosynthesis pathways, and thus these pathways should not be neglected in studies of the fertility transformation mechanism. Jasmonic acid (JA) is another vital material with important roles in the control of plant growth and development, stress responses, anther dehiscence, and the regulation of various physiological functions through rapid signal transduction pathways (León and Sánchez-Serrano 1999). JA is ubiquitous in the plant kingdom and widely distributed in the young tissues, flowers, and developing reproductive organs of plants, and it was originally isolated from the essential oil of jasmine as an odorant substance (Jiang et al. 2010). The JA biosynthetic pathway is essential for male sterility and the enzymes in this pathway have important specific roles. The key enzymes in the phenylpropanoid biosynthesis and JA synthesis pathways comprise phenylalanine ammonia-lyase (PAL), lipoxygenase (LOX), 4-coumarate: coenzyme A ligase (4CL), allene oxide cyclase (AOC), allene oxide synthase (AOS), and 12-oxo-phytodienoic acid reductase (OPR) (Ye et al. 2017). The dynamic changes in these pathways can directly affect the normal development of pollen grains.
4110S is a novel thermo-sensitive genic male sterility (TGMS) wheat line that is completely sterile at high temperatures (daily average temperature of ~ 20 °C in Zadoks growth stages 45–50) but fertile in low-temperature conditions. The crucial growth period is the later uninucleate stage (Zadoks 50). TGMS line 4110S can be applied in a two-line hybrid system to maintain male sterility via self-pollination at lower temperatures, whereas hybrid wheat seeds are produced at higher temperatures. Thus, the two-line breeding system based on TGMS line 4110S has been used in hybrid wheat breeding. In this study, we investigated the transcription profile of wheat anthers by RNA sequencing (RNA-seq) to identify the differentially expressed genes (DEGs) and some metabolic pathways related to fertility conversion. To further elucidate the molecular mechanisms responsible for fertility conversion in wheat, the possible effects of these DEGs on fertility and their biological functions were determined. The results of this study provide novel insights into the fertility conversion mechanism in TGMS wheat lines.
Materials and methods
Plant material
The TGMS wheat line 4110S used in this study was planted in 10 flower pots (30 cm high and 30 cm in diameter) at Northwest A&F University Experiment Station in Yangling (34° 29′ N, 108° 08′ E), China, during 2017–2018, and they were kept in there before they were moved into incubators. The pots containing the 4110S plants at the connectives stage (Zadoks 37) were transferred to two incubators, each with five pots, and they were exposed to two different sets of temperature conditions until the pollen production stage. The treatments were designated as ASd and ASg. The incubators had a day/night period of 14 h/10 h and a light intensity of 10000 lux, with day/night temperatures of 17 °C/15 °C for ASd and 22 °C/20 °C for ASg. The plants were cultivated until the later uninucleate stage and the anthers were then collected from the plants in the two incubators. After harvesting, the samples were frozen immediately in liquid nitrogen and stored at – 80 °C until RNA sequencing and determination of the JA contents. Anthers from the later uninucleate (Zadoks 50), binucleate (Zadoks 58), and trinucleate (Zadoks 58) stages were also preserved in formalin–acetic acid–alcohol and glutaraldehyde solution, before performing phenotypic and cytological observations (Supplementary File S1).
RNA quantification and qualification, clustering, and sequencing
The anthers obtained from spikelets at the later uninucleate stage in ASd and ASg were used to construct cDNA libraries for RNA-seq analysis, with six samples (three biological replicates for each sample) comprising ASd-1, ASd-2, ASd-3, ASg-1, ASg-2, and ASg-3 (Supplementary File S1). The total RNA was extracted according to the instruction manual provided with the RNAprep Pure Plant Kit (TIANGEN BIOTECH (Beijing) Co. Ltd., China). RNA concentrations were measured using a NanoDrop 2000 system (Thermo). RNA integrity was assessed using an RNA Nano 6000 Assay Kit with an Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). In total, 1 μg of RNA per sample was used for RNA sample preparation. Sequencing libraries were generated using a NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, USA) according to the manufacturer’s instructions and index codes were added to assign sequences to each sample. Briefly, mRNA was purified from the total RNA using poly-T oligo-attached magnetic beads. Fragmentation was conducted using divalent cations at an elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5×). First-strand cDNA was synthesized using a random hexamer primer and M-MuLV reverse transcriptase. Second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. Any remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylating the 3′ ends of the DNA fragments, the NEBNext adaptor with a hairpin loop structure was ligated before hybridization. In order to select cDNA fragments with preferred lengths of 240 bp, the library fragments were purified with the AMPure XP system (Beckman Coulter, Beverly, USA). Next, 3 μL of USER Enzyme (NEB, USA) was incubated with the size-selected, adaptor-ligated cDNA at 37 °C for 15 min, followed by 5 min at 95 °C before PCR. PCR was performed with Phusion high-fidelity DNA polymerase, universal PCR primers, and index (X) primer. Finally, the PCR products were purified (AMPure XP system) and the quality of the library was assessed with an Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v4-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina platform and paired-end reads were generated.
Quality control and comparative analysis
In order to obtain high-quality data for subsequent analyses, we filtered the raw data as follows: (a) adaptor sequences; (b) Q30 < 7.8% (Q30 means the percentage of sequences with sequencing error rates < 0.1%); and (c) N% > 10% (N means the percentage of ambiguous nucleotides). The clean reads were then mapped onto the reference genome sequence (IWGSC_RefSeq_v1.0, https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Assemblies/v1.0) (International Wheat Genome Sequencing Consortium 2014) using HISAT2.0.4 (http://ccb.jhu.edu/software/hisat2/index.shtml) (Kim et al. 2015). StringTie1.3.4b was used to assemble and quantify reads (https://ccb.jhu.edu/software/stringtie/index.shtml) (Pertea et al. 2015) (Supplementary File S1).
Screening and analysis of DEGs
FPKM values were estimated and the differential expression levels of genes in the three biological replicates were determined using the DEseq R package (1.10.1) (http://www.bioconductor.org/packages/release/bioc/html/DESeq.html) (Anders and Huber 2010), which provides statistical routines for determining the differential expression levels in digital gene expression data by using a model based on the negative binomial distribution. Fold changes ≥ 4 and FDR < 0.01 were used as the threshold values for significantly different expression levels. The P values obtained were adjusted by using Benjamini and Hochberg’s approach for controlling the FDR (false discovery rate). Genes with an adjusted P value < 0.05 were finally assigned as DEGs (Supplementary File S1).
Gene functional annotation of DEGs
Functional annotation was conducted based on the NR (ftp://ftp.ncbi.nih.gov/blast/db/), Swiss-Prot (http://www.uniprot.org/), GO (http://www.geneontology.org/), KOG (http://www.ncbi.nlm.nih.gov/KOG/), Pfam (http://pfam.xfam.org/), and KEGG (http://www.genome.jp/kegg/) databases by BLAST2.2.31 (Altschul et al. 1997). The results for new genes based on KEGG Orthology were obtained using KOBAS2.0 (Wu et al. 2006; Xie et al. 2011; Ai and Kong 2018). Alignment with the Pfam database was conducted using HMMER3.1b2 after predicting the amino acid sequences for new genes. Transcription factors were annotated with iTAK1.7 (http://itak.feilab.net/cgi-bin/itak/index.cgi) (Zheng et al. 2016) (Supplementary File S1).
Data validation and gene expression analysis by qRT-PCR
qRT-PCR validation was conducted by using the total RNA from samples ASd and ASg, which we employed for RNA-Seq analysis. Sequence-specific primers were designed using Primer Premier 5.0 software (Primer, Palo Alto, CA, USA) and tested with Primer-BLAST (Ye et al. 2017). The sequence-specific primers employed for qRT-PCR comprised the actin gene and the five selected genes listed in Supplementary Table S1. The primers were synthesized by Sangon Biotech (Shanghai) Co. Ltd (China). qRT-PCR was performed with the QuantStudioTM Real-Time PCR System (Applied Biosystems, USA) using 2× RealStar Green Power Mixture (GenStar BioSolutions (Beijing) Co. Ltd, China). The three-step cycling parameters were as follows: an initial hot start at 95 °C for 30 s, followed by 40 cycles at 95 °C for 15 s and 60 °C for 30 s. At least three technical replicates were conducted for each sample, and the relative gene expression levels were calculated using the 2–ΔΔCt method (Livak and Schmittgen 2001).
Phenotypic and cytological characteristics
Anthers were harvested at three developmental stages comprising the later uninucleate stage, binucleate stage, and trinucleate stage (Supplementary File S1). Ten anthers of each sample were fixed in glutaraldehyde solution for phenotypic observation by scanning electron microscopy (JSM-6360LV, JEOL) and the pollen wall was observed by transmission electron microscopy (JEM-1230, JEOL) (Supplementary File S1). Ten anthers for each sample at the later uninucleate stage, binucleate stage and trinucleate stage, were preserved in formalin–acetic acid–alcohol fixing solution to analyze the chromosomes by DAPI staining and determine fertility at trinucleate stage by I2-KI solution staining for 10 and 5 min, respectively (Supplementary File S1). All images were captured using an Axio Imager A2 microscope.
Determination of JA contents of anthers by ELISA
We collected 400 mg of anthers at the later uninucleate stage from ASd and ASg to measure the amounts of JA (this stage was the same as that used for RNA-seq analysis) (Supplementary File S1). The anthers were rinsed in ice-cold phosphate-buffered saline and the samples then cut into small pieces in homogenizing medium containing Tris-HCl (0.05 mol/L) and phosphate-buffered saline (pH 7.4) in an ice-cold water bath. The tissue was ground into a homogenate (10 s/time, 3–5 times) and centrifuged (3000×g, 10 min, 4 °C). Finally, the supernatant was analyzed by ELISA according to the kit manufacturer’s protocol (Sino Best Biotech (Shanghai) Co. Ltd, China). ASd and ASg were each tested three times and the data were analyzed with Excel 2010.
Results
Primary data analysis
In order to obtain an overview of the transcriptome components that are involved in the fertility conversion mechanism in 4110S, cDNA libraries were constructed from 4110S under permissive conditions (low temperatures, day/night = 17 °C/15 °C, designated as ASd) and 4110S under restricted conditions (high temperatures, day/night = 22 °C/20 °C, designated as ASg) in the late uninucleate stage (Zadoks 50) based on three biological replicates. Pearson’s correlation coefficients ranged between 0.8 and 0.994 (Fig. 1a), thereby indicating that the correlations between the sample replicates were very high. According to the clean data, the Q30 base percentage was ≥ 92.20% and the GC content was distributed from 54.54 to 55.85%. The good-quality clean reads from each sample were mapped onto the reference genome sequence IWGSC_RefSeq_v1.0 (https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Assemblies/v1.0) (International Wheat Genome Sequencing Consortium (IWGSC) 2014), where the efficiency of the comparisons ranged from 90.17 to 91.32%. The data indicated that the sequencing quality was sufficiently accurate (Table 1). Box plots can be employed to examine the degree of dispersion for the horizontal gene expression distribution in a single sample and to intuitively compare the overall gene expression levels in different samples. The box plots obtained based on the relative log reads per kilobase of transcript per million mapped reads (FPKM) values for each sample library suggested that the differences in the distributions were low among the six libraries (Fig. 1b), and thus the transcription profiles were similar. Volcano plots were generated to compare the DEGs in ASd and ASg with significant differences. Under the two different fertility conditions, genes with an adjusted fold change ≥ 4 and false discovery rate (FDR) < 0.01 were regarded as DEGs in this study. In total, 3420 DEGs were detected between ASd and ASg with 2331 upregulated genes and 1089 downregulated genes (Fig. 1c, d).

Pearson’s correlation coefficients for biological replicates in ASd and ASg (a). Box plots of relative log RPKM values based on all genes for each RNA-seq library (b). Differences in the abundance of genes between ASd and ASg (c). Differences in transcript abundance levels between ASd and ASg (d)
Functional classification of DEGs
Gene Ontology (GO) is an international standard classification system that provides a dynamic and updated standard vocabulary for describing the attributes of genes and their products in organisms. GO was used to assign the 3420 DEGs to 54 functional groups in three broad categories comprising “cellular components,” “molecular functions,” and “biological processes,” where 20 GO terms were assigned to biological processes, 17 GO terms to cellular components, and 17 GO terms to molecular functions. In the biological process category, “metabolic process” (21%) accounted for the most DEGs, followed by “cellular process” (18%) and “single-organism process” (15%). Most of the DEGs were related to the “cell part” subcategory (23%) in the cellular component category, followed by “cell” (23%) and “organelle” (19%). In the molecular function category, the majority of the DEGs were significantly enriched in the “binding” (43%) and “catalytic activity” subcategories (40%), and a relatively large number were associated with “transporter activity” (4%) (Fig. 2a, b, c and detailed information is provided in Supplementary Table S2). These results indicate that the fertility conversion mechanism may be related to many genes. GO enrichment was done for further study, oxidation-reduction process (GO:0055114) was the largest enrichment process (Supplementary Fig. S1). In “cellular components,” cytoplasmic vesicle (GO:0016023), photosystem I (GO:0009522), photosystem I reaction center (GO:0009538), chloroplast thylakoid membrane (GO:0009535) were in the top in “biological process” (Supplementary Fig. S2). Heme binding (GO:0020037), chlorophyll binding (GO:0016168), peroxidase activity (GO:0004601), sequence-specific DNA binding (GO:0003700), and serine-type endopeptidase inhibitor activity (GO:0004867) were enriched with many genes in “molecular functions” (Supplementary Fig. S3).

Gene Ontology (GO) classifications of differentially expressed genes: biological processes (a) molecular functions (b), and cellular components (c), Eukaryotic Orthologous Groups (KOG) functional classifications of annotated differentially expressed genes (d)
In total, 1494 DEGs were used to search the Eukaryotic Orthologous Groups (KOG) database to obtain functional predictions and classifications, and approximately 43.68% of the DEGs were assigned to 25 major functional categories. The R group representing “general function prediction only” comprised 260 DEGs and it was the largest group (17.40%). Genes were identified in major groups comprising “signal transduction mechanisms” (11.44%), “posttranslational modification, protein turnover, chaperones” (10.63%), “carbohydrate transport and metabolism” (9.22%), “secondary metabolites biosynthesis, transport, and catabolism” (8.14%), and “energy production and conversion” (5.47%) (Fig. 2d and detailed information is provided in Supplementary Table S3). These results demonstrated that the fertility conversion mechanism may be associated with many regulatory processes.
Identification of transcription factors
Transcription factors have essential roles in plant growth and development, morphogenesis, and metabolic regulation. We annotated 93 DEGs as transcription factors from 19 families. The largest family was MYB with 24 genes, followed by bHLH, AP2/ERF, NAC, and C2H2 with twelve, twelve, seven, and seven genes, respectively (Fig. 3 and detailed information is provided in Supplementary Table S4). The identified transcription factors suggested potential molecular mechanisms related to fertility conversion, and the results also indicated that the transcriptional regulatory network associated with fertility transformation is highly complex.

Distributions of different transcription factor families identified in ASd and ASg
Key pathways and network associated with fertility
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was employed to systematically analyze the metabolic pathways and functions for gene products and secondary metabolites as well as the biological functions of different genes in a coordinated manner. Thus, we conducted comparisons by using the KEGG database in order to obtain insights into the important biochemical pathways related to the DEGs (Fig. 4). The KEGG Orthology IDs for 651 DEGs were mapped to five categories comprising cellular processes, environmental information processing, genetic information processing, metabolism, and organismal systems, and 104 pathways (Supplementary Table S5). The highly enriched KEGG pathways were mainly related to metabolism, i.e., photosynthesis (63 genes, 9.68%) and photosynthesis-antenna proteins (63 genes, 9.68%), followed by carbon metabolism (59 genes, 9.06%), phenylpropanoid biosynthesis (50 genes, 7.68%), starch and sucrose metabolism (45 genes, 6.91%), carbon fixation in photosynthetic organisms (43 genes, 6.61%), glutathione metabolism (36 genes, 5.53%), glyoxylate and dicarboxylate metabolism (35 genes, 5.38%), phenylalanine metabolism (31 genes, 4.76%), and biosynthesis of amino acids (seven genes, 4.83%). Thus, many pathways may be involved in the regulation and control of changes in fertility.

Kyoto Encyclopedia of Genes and Genomes (KEGG) classifications of differentially expressed genes. The horizontal axis represents the gene number and the proportion of the total number of annotated genes, and the vertical axis shows the annotations of the different KEGG pathways
?>Based on the results, we analyzed three pathways associated with fertility in greater detail, which comprised the phenylpropanoid, long-chain fatty acid, and JA biosynthetic pathways. The synthesis of feruloyl-CoA proceeds via a series of reactions. In the first step, phenylalanine produces cinnamic acid under the action of the rate-limiting enzyme PAL. We found that PAL was downregulated, and p-coumaric acid, caffeic acid, and ferulic acid were synthesized. Moreover, 4CL and shikimate O-hydroxycinnamoyltransferase (HCT) were downregulated in the next reactions. Pyruvate produces acetyl-CoA via a series of reactions and the dihydrolipoamide dehydrogenase enzyme exhibited a decreasing trend in this process. In the reaction from acetyl-CoA to long-chain fatty acids, the enzymes 3-ketoacyl-CoA synthase (KCS) and 17beta-estradiol 17-dehydrogenase (KAR) were downregulated (Fig. 5a and detailed information is provided in Supplementary Table S6).

Phenylpropanoid (a) and jasmonic (b) biosynthetic pathways, and hierarchical clustering of genes encoding related enzymes (c)
In another pathway, jasmonates are first synthesized by the release of α-linolenic acid from the chloroplast membranes and a series of enzymatic reactions employ them as substrates. In our study, 13(S)-hydroperoxy-octadecatrienoic acid was formed under the action of the enzyme LOX, which had a low abundance. The product from the previous step was transformed into 12,13(S)-epoxyoctadecatrienoic acid by AOS, which was also downregulated according to our analysis. Next, 12,13(S)-epoxyoctadecatrienoic acid was converted into 12-oxo-phytodienoic acid in the plastid under the control of AOC. JA was finally synthesized in the peroxisome via three cycles of β-oxidation by OPR, and OPR was also downregulated in this process. In summary, the experimental results indicated that several enzymes, including LOX, AOX, and OPR, had low abundances during the synthesis of JA, thereby leading to decreased JA production, anther indehiscence, and male sterility (Fig. 5b and detailed information is provided in Supplementary Table S6). The important enzymes identified in this pathway in our study are shown in a heatmap in Fig. 5c.
Quantitative real-time PCR validation of candidate genes
To confirm the reliability of the transcriptome sequencing data, quantitative real-time PCR (qRT-PCR) was applied to quantify five DEGs from the key pathways based on their transcript levels, i.e., LOX2S (TraesCS7A01G246200), AOS (TraesCS4B01G237600), KCS (TraesCS1B01G387800), PAL (TraesCS1B01G122800), and OPR (TraesCS7A01G174700). The qRT-PCR results indicated that the results obtained for all of the candidate genes that we selected randomly showed a downward trend, which agreed well with the transcriptome sequencing data in the expression model (Fig. 6). Thus, the RNA-seq data were accurate, thereby validating the associations between these metabolic pathways and male fertility.

Quantitative real-time PCR validation of the RNA-Seq results for the candidate differentially expressed genes. Log2 (FC) represents the fold change in ASg relative to that in ASd
Phenotypic characteristics and microspore development
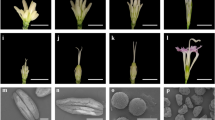
Based on the RNA-seq data analysis, we conducted further phenotypic and cytological analyses. The phenotypic characteristics of the anthers and microspores were observed during the trinucleate stage (Zadoks 58) by scanning electron microscopy and transmission electron microscopy. The upper ends of the anthers were tapered in ASg plants and they clearly differed from those in ASd (Fig. 7a, j). The exine of the microspores was rough in ASg and the baculum and tectum were disorganized, whereas the structure was regular in the ASd plants (Fig. 7i, r). Anther indehiscence and abnormal pollen exine development led to the induction of male sterility under high-temperature conditions.

Comparison of scanning electron micrograph observations, I2–KI staining, and pollen exine in ASd (a–i) and ASg (j–r) in the trinucleate stage. Anthers (a, j), outer epidermal cells (b, f, k, o), inner epidermal cells (c, g, l, p), trinucleate cells (d, h, m, q), I2–KI staining (e, n), and pollen exine (i, r). Scale bars = 1 μm (h, q), 2 μm (g, i, p, r), 10 μm (d, f, m, o), 50 μm (e, n), 100 μm (b, c, k, l), 500 μm (a, j)
We also characterized other differences in the structural features of the anthers and microspores in ASd and ASg. The outer and inner epidermis of anthers were enlarged, where the outer epidermal cells were arranged in an irregular manner and the Ubisch bodies were sparse in the inner epidermis in ASg (Fig. 7b, c, f, g, k, l, o, p). The microspores were smooth and rounded in ASd, whereas they were irregular in shape in ASg (Fig. 7d, m, h, q). Thus, ASd and ASg exhibited different phenotypic features. Moreover, fertility was confirmed by I2–KI staining, where the pollen from ASd exhibited deep and uniform staining (Fig. 7e, n), whereas the pollen from ASg was abortive and the type was stainable abortion.
In order to identify the cytological mechanism responsible for fertility conversion, 4′,6-diamidino-2-phenylindole (DAPI) staining was conducted to observe the development of the microspores in ASd and ASg. The results indicated that development was abnormal in ASg during the later uninucleate stage, where the microspores were shrunken and irregular in shapes (Fig. 8a, d). During the binucleate stage (Zadoks 58), the cytoplasm was thicker and two nuclei were apparent in ASd, as the nutrient levels started to increase. By contrast, the status of the nuclei became large in ASg (Fig. 8b, e). During the trinucleate stage, two sperm nuclei and a vegetative nucleus were observed in ASd, but the sperm nucleus was round and replaced the spindle type in ASg (Fig. 8c, f).

Staining of microspores with 4′,6-diamidino-2-phenylindole (DAPI) in ASd (a–c) and ASg (d–f) plants: later uninucleate stage (a, d), binucleate stage (b, e), and trinucleate stage (c, f). Scale bars = 50 μm
JA contents of ASd and ASg
Pathway analysis indicated that the abundance of JA was lower in ASg than ASd and the phenotypic observations indicated that no dehiscent anthers were formed by ASg. The biosynthesis of JA is of vital importance for anther development. In order to measure JA content, we applied enzyme-linked immunosorbent assay (ELISA) experiments. The results showed that the mean JA contents in ASd for three biological repeat was 17.66 μ/g, and ASg was 9.21 μ/g, according to Duncan’s new multiple range test revealed that JA contents were significantly lower in ASg than ASd (P < 0.01) (Fig. 9), and thus the lower JA contents led to the anthers failing to crack open and male sterility.

Comparison of JA contents in ASd and ASg. Data represent mean ± standard error of mean based on three biological replicates. Asterisks denote significant differences between ASd and ASg (P < 0.01, **)
Discussion
Pollen is necessary for fertility in flowering plants where it is produced by the male reproductive organs comprising anthers (Wang et al. 2017). The cytoderm is a basic component of plant cells and the pollen wall affects pollen development, and thus the fertilization process. During microspore development, the pollen wall ensures that the complete internal structure of the microspore is maintained. Thus, the leakage of microspore components from a defective pollen wall will lead to the abortion of microspores (Shi et al. 2007). Therefore, a defective pollen coat structure or lack of appropriate components such as lipids and phenolics in the pollen wall will prevent normal pollen development and eventually lead to male sterility (Preuss et al. 1993; Mayfield and Preuss 2000). The complete pollen wall structure comprises the outer and inner walls to protect the pollen grains. The exine layer is divided into the outer sexine and inner nexine (Wang et al. 2018). The proteins in this layer are derived from the tapetum in the sporophyte and they are genotype-specific. The exine layer is often yellow and sticky, where it mainly comprises sporopollenin, flavonoids, and oils (Piffanelli et al. 1998; Blackmore et al. 2007). The baculum and tectum are formed via the deposition of sporopollenin. Sporopollenin is the most important component of exine and it mainly comprises a mixture of highly resistant biopolymers, including long-chain fatty acids and phenolic compounds, which are produced via phenylpropanoid and long-chain fatty acid biosynthesis pathways (Yang et al. 2014). PAL, 4CL, and C4H are the rate-limiting enzymes in the phenylpropanoid metabolic pathway. These enzymes are located at the bifurcation of the metabolic synthesis pathway and they are the general synthetic precursors of phenolic secondary metabolites (Du et al. 2005). In our study, the downregulation of the PAL, 4CL, and HCT enzymes in the phenylpropanoid biosynthesis pathway may have led to the reduced synthesis of the intermediate product feruloyl-CoA, thereby decreasing phenolic polymer synthesis. In the long-chain fatty acid biosynthesis pathway, the key enzymes comprising KCS and KAR were downregulated, which resulted in reduced lipid polymer synthesis. Phenolic and lipid polymers are critical components of sporopollenin, and thus their inhibited synthesis affected the pollen wall and pollen development. We also observed the microspores during the trinucleate stage and found that the pollen exine was disorganized, where the distribution of sporopollenin was abnormal and this led to male sterility.
Jasmonates such as JA comprise a newly identified class of endogenous hormones that are widespread in plants, which have essential regulatory roles in plant growth, development, stress responses, and secondary metabolism. JA has two main physiological effects, as follows. First, JA is closely related to plant growth and development, such as anther dehiscence, fruit maturations, seed germination and growth, photosynthesis, organ growth and development, plant senescence, and death. Second, JA is associated with the defense system, where it acts as a signaling molecule to induce the expression of defense genes and the biosynthesis of defense chemicals by interacting with transcription factors in response to the presence of pathogenic bacteria, insect invasion, and external mechanical trauma (Creelman and Mullet 1997; Ishiguro et al. 2001; Howe and Schilmiller 2002; Li et al. 2002; Qi et al. 2011). In recent years, studies of JA mutants of Arabidopsis thaliana have also shown that JA participates in the regulation of pollen development (Gan et al. 2004). In addition, genetic analyses of Arabidopsis mutants have demonstrated the JA is involved with the splitting of anthers. The anthers of the JA-insensitive mutant coronatine insensitive1 (coi1) cannot split and it exhibits the characteristic features of male sterility, although the flowers are opened (Feys et al. 1994; Xie et al. 1998). Vegetative growth by the Arabidopsis thaliana fad3/fad7/fad8 mutants is normal initially, but they convert to male sterility during the flowering stage, where anther dehiscence does not occur and filament elongation is weak. The pollen lacks the capacity to germinate but it can grow to the trinucleate stage. In a previous study, a series of 18 carbon fatty acids and their metabolites were sprayed at the flowering stage but pollen fertility was restored only by JA, which is indispensable for normal pollen development in Arabidopsis thaliana (Mcconn and Browse 1996). The mutant form of the delayed dehiscence 1 (dde1)/opr3 gene leads to the characteristics of male sterility, with no pollen grains and failed anther dehiscence in the trinucleate stage (Sanders et al. 2000; Stintzi and Browse 2000). We found that the key enzymes comprising LOX2S, AOS, and OPR had decreased abundances in ASg. The downregulation of LOX2S might have reduced the synthesis of 13(S)-hydroperoxy-octadecatrienoic acid and catalysis by the AOS enzyme with a low abundance led to the decreased production of the intermediate 12,13(S)-epoxyoctadecatrienoic acid. Finally, the low abundance of OPR led to the decreased synthesis of JA, with effects on anther dehiscence and filament elongation. Heat stress temperatures are harmful to the growth and development of plants, and they can cause irreversible damage (Bokszczanin et al. 2013). JA signaling can promote heat resistance in wild-type Arabidopsis (Sharma and Laxmi 2016). We treated ASg at a high temperature above the optimum and ELISA showed that the JA contents were decreased, where anther dehiscence did not occur according to the phenotypic observations. The results indicated that the reduced abundances of key enzymes comprising LOX2S, AOS, and OPR decreased the production of intermediates to hinder the synthesis of JA so the anthers did not crack and male sterility occurred.
Abbreviations
- DEG:
-
differentially expressed gene
- GO:
-
Gene Ontology
- JA:
-
jasmonic acid
- TGMS:
-
thermo-sensitive genic male sterility
- EGMS:
-
environmental-sensitive genic male sterility
- PAL:
-
phenylalanine ammonia-lyase
- 4CL:
-
4-coumarate:coenzyme A ligase
- LOX:
-
lipoxygenase
- AOS:
-
allene oxide synthase
- AOC:
-
allene oxide cyclase
- OPR:
-
12-oxo-phytodienoic acid reductase
- KOG:
-
Eukaryotic Orthologous Groups
- HCT:
-
O-hydroxycinnamoyltransferase
- AOS:
-
allene oxide synthase
- qRT-PCR:
-
quantitative real-time PCR
- DAPI:
-
4′,6-diamidino-2-phenylindole
- KCS:
-
3-ketoacyl-CoA synthase
- KAR:
-
17-beta-estradiol 17-dehydrogenase
- ELISA:
-
enzyme-linked immunosorbent assay
- FDR:
-
false discovery rate
References
Ai C, Kong L (2018) CGPS: A machine learning-based approach integrating multiple gene set analysis tools for better prioritization of biologically relevant pathways. J Genet Genomics 45(9):489–504. https://doi.org/10.1016/j.jgg.2018.08.002
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
An H, Yang Z, Yi B, Wen J, Shen J, Tu J, Ma C, Fu T (2014) Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genome 15:258. https://doi.org/10.1186/1471-2164-15-258
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11(10):R106. https://doi.org/10.1186/gb-2010-11-10-r106
Blackmore S, Worrley AH, Skvarla JJ, Rowley JR (2007) Pollen wall development in flowering plants. New Phytol 174:483–498. https://doi.org/10.1111/j.1469-8137.2007.02060.x
Bokszczanin KL, Solanaceae Pollen Thermotolerance Initial Training Network (SPOT-ITN) Consortium, Fragkostefanakis S (2013) Perspectives on deciphering mechanisms underlying plant heat stress response and thermotolerance. Front Plant Sci 4:315. https://doi.org/10.3389/fpls.2013.00315
Chen H, Pan X (2001) Breeding and heterosis utilization of tropical and subtropical hybrid maize. Seed 2:57–58. https://doi.org/10.3969/j.issn.1001-4705.2001.02.028
Chen C, Chen G, Cao B, Lei J (2015) Transcriptional profiling analysis of genic male sterile–fertile Capsicum annuum reveal candidate genes for pollen development and maturation by RNA-Seq technology. Plant Cell Tissue Organ Cult 122:465–476. https://doi.org/10.1007/s11240-015-0784-5
Creelman RA, Mullet JE (1997) Biosynthesis and action of jasmonates in plants. Annu Rev Plant Physiol Mol Biol 48:355–381. https://doi.org/10.1146/annurev.arplant.48.1.355
Deng H, He Q (2017) Research and prospects of hybrid rice with strong heterosis in China. Sci Technol Rev 35:32–37. https://doi.org/10.3981/j.issn.1000-7857.2017.10.004
Dobritsa AA, Shrestha J, Morant M, Pinot F, Matsuno M, Swanson R, Møller BL, Preuss D (2009) CYP704B1 is a long-chain fatty acid omega-hydroxylase essential for sporopollenin synthesis in pollen of Arabidopsis. Plant Physiol 151:574–589. https://doi.org/10.1104/pp.109.144469
Du L, Zhang C, Zhu W, Zhang G (2005) The synthetic way and biological significance of plant secondary metabolism. J Northwest A&F Univ 20:150–155. https://doi.org/10.3969/j.issn.1001-7461.2005.03.041
Feys B, Benedetti CE, Penfold CN, Turner JG (1994) Arabidopsis mutants selected for resistance to the phytotoxin coronatine are male sterile, insensitive to methyl jasmonate, and resistant to a bacterial pathogen. Plant Cell 6:751–759. https://doi.org/10.2307/3869877
Fu Q, Cao Y, Li Y (2010) Advanced on studies and applications of photo-thermo-sensitive male sterility in wheat. J Trit Crops 30:576–580. https://doi.org/10.7606/j.issn.1009-1041.2010.03.038
Fu D, Xiao M, Hayward A, Jiang G, Zhu L, Zhou Q, Li J, Zhang M (2015) What is crop heterosis: new insights into an old topic. J Appl Genet 56:1–13. https://doi.org/10.1007/s13353-014-0231-z
Gan L, Xia K, Zhou X (2004) Involvement of jasmonates in regulation of male fertility in Arabidopsis thaliana. Plant Physiol J 40:269–274
Geng X, Ye J, Yang X, Li S, Zhang L, Song X (2018) Identification of proteins involved in carbohydrate metabolism and energy metabolism pathways and their regulation of cytoplasmic male sterility in wheat. Int J Mol Sci 19:324. https://doi.org/10.3390/ijms19020324
Howe GA, Schilmiller AL (2002) Oxylipin metabolism in response to stress. Curr Opin Plant Biol 5:230–236. https://doi.org/10.1016/S1369-5266(02)00250-9
International Wheat Genome Sequencing Consortium (IWGSC) (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345(6194):1251788. https://doi.org/10.1126/science.1251788
Ishiguro S, Kawai-Oda A, Ueda J, Nishida I, Okada K (2001) The DEFECTIVE IN ANTHER DEHISCIENCE gene encodes a novel phospholipase A1 catalyzing the initial step of jasmonic acid biosynthesis, which synchronizes pollen maturation, anther dehiscence, and flower opening in Arabidopsis. Plant Cell 13:2191–2209. https://doi.org/10.2307/3871502
Jeong HJ, Kang JH, Zhao M, Kwon JK, Choi HS, Bae JH, Lee H, Joung YH, Choi D, Kang BC (2014) Tomato male sterile 1035 is essential for pollen development and meiosis in anthers. J Exp Bot 65:6693–6709. https://doi.org/10.1093/jxb/eru389
Jiang H, Shi G (2009) Advanced search on male sterility in wheat. Biotechnol Bull 10:30–33. https://doi.org/10.13560/j.cnki.biotech.bull.1985.2009.10.027
Jiang K, Pi Y, Hou R, Tang K (2010) Jasmonate biosynthetic pathway: Its physiological role and potential application in plant secondary metabolic engineering. Bull Bot 45:137–148. https://doi.org/10.3969/j.issn.1674-3466.2010.02.001
Ju L, Zhu D, Zhang S, Zhang J, Yu Y, Liu H, Niu N, Wang J (2018) Correlation analysis between aspartic proteinase and tapetum metabolism in physiological male sterile line wheat. J Nuclear Agric Sci 32:0430–0437. https://doi.org/10.3864/j.issn.0578-1752.2017.21.002
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4):357–360. https://doi.org/10.1038/nmeth.3317
León J, Sánchez-Serrano JJ (1999) Molecular biology of jasmonic acid biosynthesis in plants. Plant Physiol Biochem 37:373–380. https://doi.org/10.1016/s0981-9428(99)80043-6
Li C, Williams MM, Loh YT, Lee GI, Howe GA (2002) Resistance of cultivated tomato to cell content-feeding herbivores is regulated by the octadecanoid-signaling pathway. Plant Physiol 130:494–503. https://doi.org/10.1104/pp.005314
Li J, Han S, Ding X, He T, Dai J, Yang S, Gai J (2015) Comparative transcriptome analysis between the cytoplasmic male sterile line NJCMS1A and its maintainer NJCMS1B in soybean (Glycine max (L.) Merr.). PLoS One 10:e0126771. https://doi.org/10.1371/journal.pone.0126771
Liu G, Xu S, Liu X (2006) Characteristics of “NEWFREE” CMS line and utilization in ornamental sunflower. Acta Agron Sin 32:1752–1755. https://doi.org/10.3321/j.issn:0496-3490.2006.11.026
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Mayfield JA, Preuss D (2000) Rapid initiation of Arabidopsis pollination requires the oleosin-domain protein GRP17. Nat Cell Biol 2:128–130. https://doi.org/10.1038/35000084
Mcconn M, Browse J (1996) The critical requirement for linolenic acid is pollen development, not photosynthesis, in an Arabidopsis mutant. Plant Cell 8:403–416. https://doi.org/10.1105/tpc.8.3.403
Meng L, Liu Z, Zhang L, Hu G, Song X (2016) Cytological characterization of a thermo-sensitive cytoplasmic male-sterile wheat line having K-type cytoplasm of Aegilops kotschyi. Breed Sci 66:752–761. https://doi.org/10.1270/jsbbs.16039
Meyer RC, Törjék O, Becher M, Altmann T (2004) Heterosis of biomass production in Arabidopsis establishment during early development. Plant Physiol 134:1813–1823. https://doi.org/10.1104/pp.103.033001
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL (2015) Stringtie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33:290–295. https://doi.org/10.1038/nbt.3122
Piffanelli P, Ross JHE, Murphy DJ (1998) Biogenesis and function of the lipidic structures of pollen grains. Sex Plant Reprod 11:65–80. https://doi.org/10.1007/s004970050122
Preuss D, Lemieux B, Yen G, Davis RW (1993) A conditional sterile mutation eliminates surface components from Arabidopsis pollen and disrupts cell signaling during fertilization. Genes Dev 7:974–985. https://doi.org/10.1101/gad.7.6.974
Qi T, Song S, Ren Q, Wu D, Huang H, Chen Y, Fan M, Peng W, Ren C, Xie D (2011) The jasmonate-ZIM-domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell 23:1795–1814. https://doi.org/10.1105/tpc.111.083261
Sanders PM, Lee PY, Biesgen C, Boone JD, Beals TP, Weiler EW, Goldberg RB (2000) The Arabidopsis delayed dehiscence1 gene encodes an enzyme in the jasmonic acid synthesis pathway. Plant Cell 12:1041–1061. https://doi.org/10.1105/tpc.12.7.1041
Sharma M, Laxmi A (2016) Jasmonates: emerging players in controlling temperature stress tolerance. Front Plant Sci 6:1129. https://doi.org/10.3389/fpls.2015.01129
Shi J, Liang WQ, Zhang DB (2007) Pollen wall development in plant. Plant Physiol J 43:588–592
Stintzi A, Browse J (2000) The Arabidopsis male-sterile mutant, opr3, lacks the 12-oxophytodienoic acid reductase required for jasmonate synthesis. Proc Natl Acad Sci U S A 9:10625–10630. https://doi.org/10.1073/pnas.190264497
Tang Z, Zhang L, Xu C, Yuan S, Zhang F, Zheng Y, Zhao C (2012) Uncovering small RNA-mediated responses to cold stress in a wheat thermosensitive genic male-sterile line by deep sequencing. Plant Physiol 159:721–738. https://doi.org/10.1104/pp.112.196048
Wang S, Lu J, Song XF, Ren SC, You C, Xu J, Liu CM, Ma H, Chang F (2017) Cytological and transcriptomic analyses reveal important roles of CLE19 in pollen exine formation. Plant Physiol 175:1186–1202. https://doi.org/10.1104/pp.17.00439
Wang K, Guo ZL, Zhou WT, Zhang C, Zhang ZY, Lou Y, Xiong SX, Yao XZ, Fan JJ, Zhu J, Yang ZN (2018) The regulation of sporopollenin genes for rapid pollen wall formation. Plant Physiol 178:283–294. https://doi.org/10.1104/pp.18.00219
Wu J, Mao X, Cai T, Luo J, Wei L (2006) KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res 34:W720–W724
Xie DX, Feys BF, James S, Nieto-Rostro M, Turner JG (1998) COl1: an Arabidopsis gene required for jasmonate-regulated defense and fertility. Science 280:1091–1094. https://doi.org/10.1016/1369-5266(88)80029-3
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li C, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:W316–W322. https://doi.org/10.1093/nar/gkr483
Xu Y, Kang H (2001) Research progress on the two-lines hybrid wheat of photo-thermo-sensitivity. Shananxi J Agric Sci 01:28–29. https://doi.org/10.3969/j.issn.0488-5368.2001.09.012
Yang X, Wu D, Shi J, He Y, Pinot F, Grausem B, Yin C, Zhu L, Chen M, Luo Z, Liang W, Zhang D (2014) Rice CYP703A3, a cytochrome P450 hydroxylase, is essential for development of anther cuticle and pollen exine. J Integr Plant Biol 56:979–994. https://doi.org/10.1111/jipb.12212
Yang X, Geng X, Liu Z, Ye J, Zhang L, Song X (2018) A sterility induction trait in the genic male sterility wheat line 4110S induced by high temperature and its cytological response. Crop Sci 58:1–11. https://doi.org/10.2135/cropsci2017.12.0714
Ye J, Duan Y, Hu G, Geng X, Zhang G, Yan P, Liu Z, Zhang L, Song X (2017) Identification of candidate genes and biosynthesis pathways related to fertility conversion by wheat KTM3315A transcriptome profiling. Front Plant Sci 8:449. https://doi.org/10.3389/fpls.2017.00449
Zhang W (2000) Improving rape yield by heterosis. Gansu Agric 2:39–40. https://doi.org/10.15979/j.cnki.cn62-1104/f.2000.02.016
Zheng Y, Jiao C, Sun H, Rosli HG, Pombo MA, Zhang P, Banf M, Dai X, Martin GB, Giovannoni JJ, Zhao PX, Rhee SY, Fei Z (2016) iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol Plant 9:1667–1670. https://doi.org/10.1016/j.molp.2016.09.014
Acknowledgments
We thank Beijing BioMarker Company for their help with high-throughput sequencing. The data from this study was deposited in NCBI Sequence Read Archive under accession SRA: SRP211924. The line 4110S was presented by Dr. P.H. Dong from the College of Agronomy, Henan University of Science and Technology, China.
Funding
This study was supported by the National Natural Science Foundation of China (31771874) and the Program in Science and Technology of Yangling State Demonstration Zone of Agricultural High-tech Industries (2018NY-19).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experiments were carried out with the approval of Northwest A&F University.
Competing interest
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary materials
Supplementary Table S1
Sequence-specific primers used for qRT-PCR (PDF 117 kb)
Supplementary Table S2
Gene Ontology (GO) classifications of differentially expressed genes (PDF 174 kb)
Supplementary Table S3
Eukaryotic Orthologous Groups (KOG) functional classifications of annotated differentially expressed genes (PDF 158 kb)
Supplementary Table S4
Transcript facror IDs and corresponding families (PDF 198 kb)
Supplementary Table S5
KEGG pathways and DEGs number (PDF 133 kb)
Supplementary Table S6
Genes and KEGG terms in networks (PDF 116 kb)
Supplementary Fig. S1
The GO enrichment of biological processes (PDF 578 kb)
Supplementary Fig. S2
The GO enrichment of cellular components (PDF 486 kb)
Supplementary Fig. S3
The GO enrichment of molecular functions (PDF 356 kb)
Supplementary File S1
The detailed experimental methods. (PDF 135 kb)
Rights and permissions
About this article

Cite this article
Yang, X., Ye, J., Zhang, L. et al. Blocked synthesis of sporopollenin and jasmonic acid leads to pollen wall defects and anther indehiscence in genic male sterile wheat line 4110S at high temperatures. Funct Integr Genomics 20, 383–396 (2020). https://doi.org/10.1007/s10142-019-00722-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-019-00722-y