
Abstract
Rhizoctonia solani AG1-IA is a soil-borne necrotrophic pathogen that causes devastating rice sheath blight disease in rice-growing regions worldwide. Sclerotia play an important role in the life cycle of R. solani AG1-IA. In this study, RNA sequencing was used to investigate the transcriptomic dynamics of sclerotial development (SD) of R. solani AG1-IA. Gene ontology and pathway enrichment analyses using the Kyoto Encyclopedia of Genes and Genomes (KEGG) were performed to investigate the functions and pathways of differentially expressed genes (DEGs). Six cDNA libraries were generated, and more than 300 million clean reads were obtained and assembled into 15,100 unigenes. In total, 12,575 differentially expressed genes were identified and 34.62% (4353) were significantly differentially expressed with a FDR ≤ 0.01 and |log2Ratio| ≥ 1, which were enriched into eight profiles using Short Time-series Expression Miner. Furthermore, KEGG and gene ontology analyses suggest the DEGs were significantly enriched in several biological processes and pathways, including binding and catalytic functions, biosynthesis of ribosomes, and other biological functions. Further annotation of the DEGs using the Clusters of Orthologous Groups (COG) database found most DEGs were involved in amino acid transport and metabolism, as well as energy production and conversion. Furthermore, DEGs relevant to SD of R. solani AG1-IA were involved in secondary metabolite biosynthesis, melanin biosynthesis, ubiquitin processes, autophagy, and reactive oxygen species metabolism. The gene expression profiles of 10 randomly selected DEGs were validated by quantitative real-time reverse transcription PCR and were consistent with the dynamics in transcript abundance identified by RNA sequencing. The data provide a high-resolution map of gene expression during SD, a key process contributing to the pathogenicity of this devastating pathogen. In addition, this study provides a useful resource for further studies on the genomics of R. solani AG1-IA and other Rhizoctonia species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is the staple food for nearly half of the world population, especially in Asia (Kuanar et al. 2017). Rice sheath blight (RSB), caused by Rhizoctonia solani AG1-IA (teleomorph Thanatephorus cucumeris), is a devastating disease affecting rice crops throughout the world (Moni et al. 2016). In recent years, RSB has become one of the most prominent diseases in rice-growing regions of China due to the widely adopted high-yielding semidwarf rice cultivars, heavy use of nitrogen-based fertilizers, and an increasing number of sclerotia found in field soils (Wang et al. 2015). In 2016, RSB affected ~ 18 million ha of paddy-irrigated rice in China leading to significant economic losses.
In nature, R. solani does not produce any asexual spores and survives mainly as its mycelial and sclerotial forms in crop residues and soil (Damicone et al. 1993; Kwon et al. 2014; Feng et al. 2017). The hard dormant sclerotia can survive in soil without a host for 8–10 years (Willetts and Bullock 1992), and serve as the primary source of infection for rice sheath blight (Feng et al. 2017; Srinivas et al. 2014). Under favorable conditions, the sclerotia of R. solani germinate to form hyphae in soil, which penetrate into the sheath, and can grow both inside the plant and on the surface. The fungus causes water-soaked lesions on the sheath and leaves, and initial lesions may expand to envelop the whole plant as the disease progresses. At later stages of RSB, vegetative hyphae aggregate into clumps of mycelia, eventually developing into mature sclerotia, which fall into the soil. The fallen sclerotia germinate causing secondary infections, or may remain dormant in soil until the following season (Taheri and Tarighi 2011,) as such playing an important role in the life cycle of RSB.
The morphological events of sclerotial development (SD) have been well studied, through both microscopic observations and via analyses of environmental cues (Brown and Wyllie 1970; New et al. 1984; Sarrocco et al. 2006; Willetts and Bullock 1992). Four types of sclerotia exist and are roughly classified based on the extent of inter-tangling and degree of aggregation. In general, a three-layered structure is typical of most sclerotia: the nutrient-rich center is known as the medulla, the middle layer is a thick-walled tissue-like cortex, and the surface layer is a sheath-like structure that accumulates large amounts of melanin (Townsend and Willetts 1954; Willetts and Bullock 1992). The structure of R. solani sclerotia is distinct, and often referred to as a primitive rind type structure with dark outer cells. The R. solani AG1-IA sclerotia are formed initially from compact masses of monilioid cells, which then enlarge by irregular branching of the mycelium, followed by intercalary septation, and swelling of the hyphae (Willetts and Bullock 1992). During the developmental and maturation stages, the surfaces of R. solani AG1-IA sclerotia become heavily melanized due to increasing phenol oxidase activity (Sumner 1996). Several environmental cues have been associated with the onset of sclerotia formation, such as nutrient depletion, light intensity and quality, temperature, pH, the presence of antioxidants, and fluctuations in aeration (Lu et al. 2016; Maurya et al. 2010; Ritchie et al. 2009; White 1976). However, little is known about the molecular mechanisms involved in R. solani AG1-IA sclerotial metamorphosis, in particular, changes in gene expression at the transcriptome level.
The first step to elucidating the molecular mechanisms underlying sclerotial development in R. solani AG1-IA is global gene expression analysis. RNA sequencing is a powerful technology providing an accurate estimation of gene expression enabling the identification of coding sequences, gene function, alternative splicing, and metabolic pathways (Finotello and Di 2015; Trapnell et al. 2010; Wang et al. 2009, 2016). RNA-Seq is progressively providing greater knowledge of the development of pathogenic fungi (Daly et al. 2016; Duressa et al. 2013; Wang et al. 2014; Xiong et al. 2014; Xu et al. 2011).
To decipher the molecular mechanisms of SD in R. solani AG1-IA, we sequenced and de novo assembled the R. solani AG1-IA transcriptome using the reference genome sequence of R. solani AG1-IA (Rice Sheath Blight Pathogen Genomics Database http://genedenovoweb.ticp.net:81/rsia/index.php) (Chen et al. 2016). In this study, we uncover developmental stage-specific gene expression patterns during sclerotial development and identify many differentially expressed genes associated with the developmental process of R. solani AG1-IA. Bioinformatics analyses were conducted to analyze 15,100 unigenes and global profiles of gene expression in three identified SD stages in R. solani AG1-IA. Based on the results, we have identified several significant differentially expressed genes that reveal the major metabolic processes and signaling pathways during sclerotial metamorphosis in R. solani AG1-IA. By elucidating the molecular mechanisms governing SD in R. solani AG1-IA, novel strategies can be designed to control Rhizoctonia disease, not only for the RSB pathogen but also for related pathogenic strains and other sclerotia producing Rhizoctonia spp.
Methods
Fungal strain, growth conditions, and development stages
The virulent GD-118 strain of R. solani AG-1 IA was collected from Guangdong province in China. Cultures of strain GD-118 were grown on potato dextrose agar (PDA) overlaid with a 9-cm cellophane membrane, and incubated at 28 °C and 65% relative humidity with a 12-h photoperiod. Samples were harvested at three distinct stages of SD, 36 h (mycelium stage (MS)), 3 days (sclerotia initial (SI)), and 14 days (sclerotia maturation, (SM)). Mycelia were scraped from the Petri dish and sclerotia collected with forceps. Newly harvested samples were frozen in liquid nitrogen and stored at − 80 °C until use.
The morphological characteristics of R. solani AG-1 IA at each stage were examined using light microscopy (LM) and transmission electron microscopy (TEM). For TEM, samples (MS, SI, and SM) were soaked in a fixative solution containing 4% (w/v) glutaraldehyde and 3% (w/v) paraformaldehyde for 22 h, post-fixed in 2% osmium tetroxide, and dehydrated in an ethanol series. Subsequently, the samples were embedded in Epon 812 resin (TAAB Laboratories Equipment, Berks, UK) and polymerized at 60 °C for 48 h. Thin sections were cut, stained with ethanolic uranyl acetate and lead citrate, and imaged using a Tecnai 12 transmission electron microscope (Philips, Netherlands), according to previously described protocol (Chen et al. 2015). For LM, samples were directly examined on an Eclipse 80i light microscope (Nikon, Japan).
RNA extraction
Extraction of total RNA from mycelia and sclerotia was performed using a Fungal RNA Kit (Omega, USA) according to the manufacturer’s instructions. Extracted RNA was then treated with DNase I to remove any genomic DNA and purified using a MicroElute RNA Clean-up Kit (Omega, USA). RNA quality and yield were measured using gel electrophoresis and spectrophotometry (Thermo Fisher Scientific, USA). RNA integrity was further assessed using a 2100 Bioanalyzer (Agilent Technologies, USA). Two biological replicates for each sample were used to prepare libraries.
Library preparation, sequencing, and data analysis
cDNA libraries were constructed using the Illumina TruSeq RNA Sample Prep Kit (Illumina, USA). To make short fragments, mRNA was isolated using Oligo (dT) beads and mixed with fragmentation buffer. First strand cDNA was synthesized from 1 μg mRNA, 1 μL Oligo (dT) 18 primer (0.1 μg/μL), and 1 μL M-MuLV Reverse Transcriptase (RNaseH−, 200 U/μL). Subsequently, second strain cDNA was synthesized using 1 μL DNA polymerase I (5 U/uL), 0.5 μL RNase H (30 U/uL), and 4 μL 0.1 mM dNTPs. To obtain 150- to 200-bp-length cDNA fragments, ds-cDNA was purified using AMPure XP beads (Beckman Coulter, Beverly, USA). The resulting cDNA fragments were ligated with adaptors and amplified using Phusion® High-Fidelity DNA polymerase.
The products of PCR, approximately 200 bp in length, were purified by gel electrophoresis, and cDNA quality was assessed using the Agilent 2100 Bioanalyzer. Index-coded samples were clustered with the TruSeq PE Cluster Kit v3-cBot- HS (Illumina, USA) according to the manufacturer’s recommendations. Finally, six libraries were sequenced using HiSeq™ 2000 (Illumina, USA)) and 100-bp paired-end reads were generated. For data analysis, quality of raw data was assessed using Fastqc (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and excess adaptors and low-quality reads were removed. The high-quality reads were then used in downstream analyses and calculated based on Q20, Q30, GC-content, and N values. Q20 and Q30 represented the percentage of sequences with sequencing error rate lower than 1 and 0.1%, respectively. N percentage is the percentage of nucleotides which could not be sequenced.
High-quality reads were mapped to the reference genome of R. solani AG1-IA (Trapnell et al. 2009) using Tophat (version 2.1.1) with default options. Individual transcripts were assembled and gene expression quantified with per kilo-base transcript per million mapped reads (FPKM) in Cufflinks (version 2.2.1) using the default parameters (Mortazavi et al. 2008; Trapnell et al. 2010). The transcripts of each sample were then merged in Cuffmerge, and DEGs were identified using Cuffdiff with FDR < 0.01 (Trapnell et al. 2012).
Gene ontology and functional annotation
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) were annotated using the GO database (www.geneontology.org) and KEGG database (http://www.genome.jp/kegg/), and functional annotation, classification, and enrichment analyses were conducted using the BLAST2GO software (https://www.blast2go.com), based on the default parameters. Gene conservation was checked by sequence similarity of unigene sequences to the genome of R. solani AG1-IB, R. solani AG3, and R. solani AG8 using blast+ (v 2.7.1)(Altschul et al. 1990). All the genome files were downloaded from NCBI bioprojects (PRJNA73133, PRJNA187548, and PRJNA187548). Blast results were filtered for 80% sequence identity. Comparative transcriptomic analysis was performed using the gene expression values from RSIADB (Chen et al. 2016; Zheng et al. 2013). Hierarchical clustering using Pearson’s correlation, principal component analysis (PCA), heat map diagrams, Venn diagrams, and Short Time-series Expression Miner (STEM) analysis were performed using OmicShare tools, a free online platform for data analysis (www.omicshare.com/tools).
Quantitative real-time reverse transcription PCR
To validate gene expression, quantitative real-time reverse transcription PCR (qRT-PCR) was performed. DNase-treated total RNA was transcribed to cDNA using the PrimerScript RT reagent Kit (TaKaRa, Japan) according to the manufacturer’s instructions. cDNA was amplified in an Illumina ECO real-time PCR system (Illumina) using SYBR Premix Ex Taq II (Tli RNaseH Plus) (TaKaRa). Primers are listed in Table S3. Relative gene expression levels were normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and calculated using the 2−ΔΔCT method (Livak and Schmittgen 2001). All assays were performed in triplicates and repeated at least three times. Statistical significance was assessed using analysis of variance (ANOVA) in SPSS (version 13.0) with p < 0.05 considered statistically significant.
Availability of supporting data
The raw reads generated from sequencing of all six libraries were deposited at the National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/Traces/sra/) with accession number SRA303035 under the BioProject PRJNA297820.
Results
Microscopic analysis of sclerotial metamorphosis
The dynamic processes involved in SD of R. solani AG-1 IA were investigated using LM and TEM, to illuminate mycelia structural and cellular changes associated with gene expression at the transcriptome level. Three stages, MS, SI and SM, were selected for microscopic examination based on the phenotypic changes observed during sclerotial morphogenesis (Fig. 1). At the MS stage, the petri dish was covered with undifferentiated hyphae. Observation of the LM stage showed sparsely septate hyphae with right-angled branching, and the TEM stage showed typical oval-shaped hyphae with no melanized particulate matters. Subsequently, vegetative hyphae aggregated at the SI stage and formed interwoven hyphal masses, which were thin-walled and loosely arranged. The hyphal branches became shorter with irregular melanized particulate matter in the cell. The MS stage was identified by observation of a thickened cell wall surrounded by melanin. Hyphae adhered to each other and the cell walls became thick and compact with heavy accumulation of melanin, as observed by TEM.

Sclerotial development stages of R. solani AG1-IA.
a SD of R. solani AG1-IA on PDA plate. b Representative light microscope images of mycelia and sclerotia. c Transmission electron microscope images of mycelia and sclerotia
Sequencing and de novo assembly of R. solani AG1-IA transcriptome
In this study, six RNA libraries, derived from three samples (MS, SI, and SM) of R. solani AG1-IA, were sequenced. After removing the excess adaptors and low-quality reads, a total of 318,851,506 reads were obtained from six libraries with a Q20 percentage of 98 and Q30 percentage of 94. An average of 53,141,918 reads were obtained for each sample, ranging from 49,746,554 to 56,219,492 (Table 1).
The R. solani AG1 IA genome assembly and the 10,590 predicted genes of this strain were used as a reference sequence (Rice Sheath Blight Pathogen Genomics Database, http://genedenovoweb.ticp.net:81/rsia/index.php) (Chen et al. 2016). In total, 75.0% of the total reads (238,485,048 reads) from all six samples aligned with the genome of R. solani AG1-IA, with less than 2 bp mismatches (Table S1).
The statistical results of the de novo assembly are shown in Table 2. A total of 15,100 unigenes were assembled, with an average of 2391 bp and an N50 of 4257 bp. A total of 14,573 unigenes (≥ 200 bp) of sufficient quality were analyzed by functional annotation. Among them, 5977 unigenes were longer than 2000 bp (Fig. S1).
To analyze the coding sequences (CDS), predicted proteins, and gene annotations, 14,573 unigenes were aligned against the NCBI non-redundant (Nr) database using the BLAST2GO software. Protein functions were annotated with top-scoring similar proteins. In total, 13,137 (90.1%) significant BLAST hits (E value ≤ 10−5) were obtained. The size distributions of CDS and predicted proteins are illustrated in Fig. S2.
To illustrate similarities between gene expression profiles, Pearson correlation was used to calculate the similarity of paired samples for 12,575 DEGs using FPKM. Samples exhibiting a similar gene expression pattern were clustered together based on the Pearson correlation. Analysis revealed SI and SM stages share the most similar gene expression profiles, while the expression patterns during MS were more distinct. Principal component analysis (PCA) was also performed on 12,575 DEGs and showed that PC1 explains 56.7% of the variance and PC2 explains 43.4%. Additionally, the gene expression profiles of the SI and SM stages clustered together, whereas the MS was more distinct. Therefore, the PCA results were in agreement with the Pearson correlation (Fig. 2).

Correlation of gene expression profiles based on three sequencing libraries. a Pearson correlation heat map. b Principal component analysis plot generated from expression data of 12,575 DEGs showing transcriptome differences among three sequencing libraries by OmicShare tools (www.omicshare.com/tools)
Annotation of R. solani AG1-IA transcript sequences
Using the Blast2GO software, GO terms and enzyme commission (EC) numbers were assigned to 14,573 unigenes. A total of 2889 unigenes were assigned to 1234 GO terms for describing biological processes, molecular functions, and cellular components. Of them, 75.8% (2189) were assigned EC numbers. To plot the GO annotations, the WEGO software (http://wego.genomics.org.cn) was used (Fig. 3). Furthermore, 2889 unigenes were categorized into 12 molecular functional groups, among which the dominant terms were “binding,” “catalytic,” “transporter,” and “structure molecule.” For cellular components, 36 groups were assigned and the dominant terms were “cell,” “organelle,” “intracellular,” and “membrane.” Among 18 biological process groups, the majority of terms were “cellular process,” “metabolic process,” “localization,” and “response to stimulus.”

Histogram of gene ontology (GO) classification. A total of 2889 unigenes were enriched into 66 terms from three ontologies, including cellular components, molecular function, and biological processes. The right y-axis represents the number of unigenes. The left y-axis denotes the unigenes in a specific category as a percentage of the main category.
The unigenes were analyzed with the COG database for functional prediction and classification. A total of 12,745 sequences were assigned to 24 COG categories (Fig. 4). Among the 24 categories, “general function prediction only” represented the largest group (2442 unigenes; 19.2%), followed by “amino acid transport and metabolism” (1063 unigenes; 8.34%), “posttranscriptional modification, protein turnover, chaperones” (923 unigenes; 7.24%), “energy production and conversion” (849 unigenes; 6.66%), and “carbohydrate transport and metabolism” (826 unigenes; 6.48%). The smallest groups were “cell motility” (17 unigenes; 0.133%) and “nuclear structure” (3 unigenes; 0.023%).

Orthologous groups of proteins (COG) annotation for putative proteins. A total of 12,745 sequences had significant homology to those in COG database and were functionally assigned to 24 molecular categories.
To identify the main biological pathways active during SD of R. solani AG1-IA, pathways with a Q value ≤ 0.01 were considered significantly enriched in DEGs. In total, 241 DEGs were assigned to 47 KEGG pathways (Table S2). The top enrichment pathways included biosynthesis of ribosomes (46 DEGs; 11.86%), limonene, and pinene degradation (18 DEGs, 4.64%) and polycyclic aromatic hydrocarbon degradation (17 DEGs; 4.38%). These results are useful for the investigation of specific mechanisms in SD of R. solani AG1-IA.
Gene expression patterns during the stages of SD
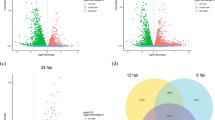
A total of 12,575 DEGs were identified, and 34.62% (4353) of them were significantly expressed (FDR ≤ 0.01 and |log2Ratio| ≥ 1) during SD of R. solani AG1-IA. Subsequently, the 4353 DEGs with significant levels of expression were analyzed via pairwise comparisons of different samples. The results showed that 2681 unigenes were significantly upregulated and 2379 were significantly downregulated, and were thus considered as candidate genes involved in sclerotial metamorphosis. The largest number of significantly upregulated (1657) and downregulated (1491) genes were seen for the SI stage compared to MS; however, 744 unigenes were upregulated and 742 downregulated in the SI compared to SM stage (Fig. 5a). Only 283 overlapping DEGs were shared across the whole developmental process of sclerotia (Fig. 5b), in which the “catalytic activity” was the top represented functional group.

Gene expression patterns of DEGs during Rhizoctonia solani AG-1 IA sclerotial development. a Statistical column diagram of the upregulated and downregulated unigenes based on pairwise comparisons of samples. The following comparisons were performed: MS versus SI, MS versus SM, and SI versus SM. b Venn diagram illustrating number of significant DEGs based on pairwise comparisons of samples. Separate and overlapping expression between samples is shown. c Heat map of global gene expression levels and hierarchical clustering for each of the three stages. All genes differentially expressed in at least one sample were included (red, upregulation; green, downregulation; black, moderate expression)
To view the overall gene expression pattern, hierarchical clustering was generated based on the log10FPKM value of significantly expressed DEGs. In general, four groups of DEGs demonstrated significant expression patterns: DEGs in groups I and II were upregulated at the SI stage; DEGs in group III were downregulated during the SD process; and group IV exhibited a mostly continuous upregulation trend (Fig. 5c).
The Short Time-series Expression Miner (STEM) program was used to delineate 74.9% (9418) of 12,575 DEGs into eight profiles (Fig. S3). Profile 0 (877 DEGs) has a sustained upward trend at the SI and SM stages, while profile 7 (836 DEGs) displayed a sustained downward trend. The profile 2 (1828 DEGs) gene clusters had a “down-up” trend across three stages, while profile 5 (1048 DEGs) showed an “up-down” expression trend. Profiles 3 (1217 DEGs) and 4 (1218 DEGs) had no change at the early stage, but were either down- or upregulated at a later stage. Profile 1 (1210) showed a downregulation trend and profile 6 (1184 DEGs) was upregulated at the SI stage, with no change at the final stage.
Functional categorization of DEGs involved in SD
Forty-eight DEGs were analyzed in silico and the gene functions, gene conservation, and comparative transcriptomic analysis were conducted, to understand any potential role in SD. These included DEGs coding for secondary metabolite biosynthesis and transport proteins, as well as coding for melanin biosynthesis, ubiquitin and autophagy, cell cycle/cytoskeleton, carbohydrate metabolism, transcription factor, and reactive oxygen species metabolism (Fig. 6). Details of the DEGs in each group are described below.

Gene expression heat map of DEGs involved in Rhizoctonia solani AG1-IA sclerotial development
Gene conservation analysis
Sequence similarity results showed that 70% (34) of DEGs were conserved in different AGs (Fig. S4). Among them, 13 DEGs co-existed in AG3, AG8, and AG1-IB; nine DEGs co-existed in AG8 and AG1-IB; and two DEGs co-existed in AG1-IB and AG3. In addition, nine DEGs shared high gene similarity with AG1-IB, and only one DEG shared high gene similarity with AG8. No specific DEG shared high gene similarity in AG3. From the Venn diagram, it is evident that a vast majority of DEGs were conserved in AG1-IB, which is a sub-group of R. solani AG1.
Secondary metabolite biosynthesis, transport proteins
To determine the expression profiles of DEGs encoding enzymes involved in secondary metabolic pathways and transport proteins at each of the three stages, FPKM values were calculated for four cytochrome P450-related genes, CYP (cytochrome P450; TCONS_00010059), P450s (cytochrome P450 monooxygenase; TCONS_00004458), CYPOR (cytochrome P450 oxidoreductase; TCONS_00002276), and one Vel (velvet; TCONS_00012102), as well as four transport protein-related genes, ABC (ATP-binding cassette; TCONS_00014550), ABC transporter protein (TCONS_00007454), IMP (integral membrane protein; TCONS_00004444), and MFS transporter (major facilitator superfamily transporter; TCONS_00009629). The results demonstrate that most DEGs exhibit a similar upregulation profile at the SI stage; however, expression tends to decrease at the SM stage, suggesting secondary metabolism and transporter proteins are actively involved in sclerotial formation.
Melanin biosynthesis
A major change that occurs during SD of R. solani AG1-IA is sclerotial color transition from white to dark, associated with the accumulation of melanin. Therefore, we calculated the FPKM values of unigenes encoding for enzymes associated with melanin biosynthesis. Four genes were identified by our data mining analysis, including PKS (polyketide synthetase; TCONS_00005501), LAC1 (laccase-1; TCONS_00006208), TYR (tyrosinase; TCONS_00007083), and PKC (polyketide cyclase; TCONS_00008578). The TYR expression profile was highly expressed at the SI stage and significantly downregulated at the SM stage. Moreover, expression of the PKC gene dramatically increased at the SM stage compared with MS and SI. These profiles were consistent with phenotypic observations of SD, in which melanin was dramatically accumulated at the SM stage (Fig. 6). In contrast, PKS and LAC1 homologs were downregulated at the MS, in comparison to the SI and SM stages.
Ubiquitin and autophagy
In this study, DEGs involved in the ubiquitin-protein degradation pathway, including Ub (ubiquitin; TCONS_00003960) and USP (ubiquitin-specific protease; TCONS_00008189), were highly expressed at the SM stage, whereas E3 (ubiquitin-protein ligase; TCONS_00008527) showed a trend of upregulation at the SI stage, and then dramatically decreased at the SM stage. Interestedly, during the process of autophagy, five DEGs encoding for proteins Vps8 (vacuolar protein; TCONS_00011673), ATG13 (TCONS_00003071), ATG16 (TCONS_00013298), ATG2 (TCONS_00003033), and STPKs (serine/threonine protein kinase; TCONS_00001409) showed a consistent trend, and relatively high expression levels were detected at the SI stage, which then dramatically decreased at the SM stage. These findings suggest a potential role for ubiquitin and autophagy related genes in SD of R. solani AG1-IA.
Cell cycle/cytoskeleton, transcription factor
Nine DEGs related to cell cycle/cytoskeleton and transcription factors were identified.
At the SI stage, one of the genes encoding a plasma membrane fusion protein was found to be strongly expressed, and genes encoding CMP (cellular morphogenesis protein; TCONS_00014075) and WD40 (TCONS_00001320) were only slightly expressed at the SI stage and the expression levels remained stable at the SM stage compared to the MS. Two DEGs encoding ACOP (actin cytoskeleton organization protein; TCONS_00013740) and spo4 (cell cycle protein kinase spo4; TCONS_00013288) showed continuous upregulation at the SI and SM stages. Four transcription factor-related genes were differentially expressed at the SI and SM stages, including btf3 (transcription factor btf3; TCONS_00004010), TIF-IA (transcription initiation factor; TCONS_00004309), NRG1 (transcriptional regulator NRG1; TCONS_00013547), and zpr1 (zinc-finger protein; TCONS_00012012). Moreover, TIF-IA and NRG1 were significantly increased at the SI stage and decreased at the SM stage. Slight expression of btf3 was observed at the SI stage, and remained stable at the SM stage. However, the expression of zpr1 showed a “down-up” trend compared to the MS stage.
Carbohydrate metabolism
In this study, we found genes encoding glycosyl hydrolase family proteins (TCONS_00004363, TCONS_00005255, and TCONS_00004868), polyscaccharide lyase family proteins (TCONS_00005607 and TCONS_00011017), and carbohydrate esterase (TCONS_00014139) had similar expression profiles, with higher levels at the SI stage dramatically decreasing at the SM stage. Three genes involved in PL3 (polysaccharide lyase family 3 protein, TCONS_00008054), TKL1 (transketolase1; TCONS_00013408), and HK1 (hexokinase; TCONS_00012962) were dramatically downregulated, suggesting the pathways were suppressed during SD. At the SM stage, a gene encoding PK (pyruvate kinase) was significantly upregulated.
Reactive oxygen species metabolism
Based on GO annotation, genes involved in antioxidant functions and response to stimuli were significantly expressed during the SD of R. solani AG1-IA. To further study the expression of DEGs involved in oxidative stress metabolism, nine DEGs involved in the metabolism of reactive oxygen species were examined. Three NADPH-related genes, Nox (NADH oxidase; TCONS_00009009), NoxR (NADPH oxidase regulator; TCONS_00004890), and NFR (NADPH-ferrihemoprotein reductase; TCONS_00010386), as well as MEP (metalloendopeptidase; TCONS_00005543) and GR (glutathione reductase; TCONS_00013972), were highly expressed at the MS and dramatically decreased at the SI and SM stages, whereas CAT (catalase, TCONS_00002674), OXNAD (oxidoreductase FAN/NAD-binding protein; TCONS_00013635), HSP (heat shock protein, TCONS_00004695), and GST (glutathione S-transferase, TCONS_00013585) were expressed in a constitutive manner, detected at relatively low expression levels at the MS and SI stage, but dramatically expressed at the SM stage. These findings indicate that oxidative stress may play an important role during SD.
Comparative transcriptomic analysis
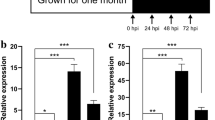
In order to reveal the gene expression profile of 48 DEGs during the six stages of progression of RSB, heat map was generated, which has two distinct clades. Twenty-eight DEGs were upregulated and 20 DEGs were downregulated (Fig. 7). Among 28 upregulated DEGs, four of them showed highest expression level at 48 h, 12 of them consistently high expressed at 72 h, eight DEGs increased at 24 h, peaked at 32 h and declined significantly at 48 h. Among 20 downregulated DEGs, 14 DEGs were highly expressed at 10–18 h and six DEGs showed highest expression level at 24–32 h.

Gene expression heat map of DEGs involved in the disease progression of rice sheath blight disease. All genes differentially expressed in at least one sample were included (red, upregulation; green, downregulation; black, moderate expression)
qRT-PCR validation
To validate the expression patterns obtained using RNA-seq, 10 unigenes showing different expression profiles during sclerotial metamorphosis were chosen for qRT-PCR analysis (Table S3). The unigenes encoded Velvet domain-containing protein (TCONS_00012102), uncharacterized protein (TCONS_00006512), small heat shock protein C4 (TCONS_00006034), hypothetical protein (TCONS_00011708), alpha,alpha-trehalose-phosphate synthase (TCONS_00007765), ZZ domain-containing protein (TCONS_00003428), aldo/keto reductase (TCONS_00006280), arm domain-containing protein (TCONS_00009538), cAMP-independent regulatory protein pac2 (SOD; TCONS_00013149), and pyridoxal-phosphate-dependent-enzyme family (TCONS_00015096) (Fig. 8). The results of qRT-PCR showed the expression profiles of 10 DEGs were well aligned to those of RNA-seq.

DEGs during Rhizoctonia solani AG-1 IA sclerotial development. a TCONS_00012102. b TCONS_00006512. c TCONS_00006034. d TCONS_00011708. e TCONS_00007765. f TCONS_00003428. g TCONS_00006280. h TCONS_00009538. i TCONS_00013149. j TCONS_00015096. Data presented as mean ± one standard error of three replicates (qRT-PCR). Columns with different letters are significantly different (p < 0.05)
Discussion
Next-generation sequencing has become an important tool for studying the genome of a variety of biological organisms. RNA-seq is an efficient and economical approach for providing comprehensive sequencing data in a relatively short timeframe and has been applied in numerous microbial studies. Here, we compared the transcriptomes of R. solani AG1-IA during three different stages of SD (i.e., MS, SI, and SM). The genes differentially expressed during SD of R. solani AG1-IA, the causal agent of RSB, were identified in order to further elucidate the molecular mechanisms involved in SD. RNA-seq was applied to identify DEGs, and the genome sequence of R. solani AG1-IA was used as a reference sequence for in silico functional annotation. Previous studies have provided an understanding of the physiology and biochemistry of sclerotial morphogenesis in R. solani AG1-IA (Georgiou and Petropoulou 2001; Hu et al. 2017). In this paper, we have used RNA-seq and the R. solani genome to uncover some of the molecular mechanisms involved in SD of R. solani AG1-IA. Among the six transcriptomes, 12,575 unigenes were differentially expressed, and 34.62% of them were significantly differentially expressed (FDR ≤ 0.01 and |log2Ratio| ≥ 1). In comparison to the 258 transcript-derived fragments obtained using the cDNA-AFLP method (Shu et al. 2015) and 28 expressed sequence tags via the SSH approach (Chen et al. 2012), RNA-Seq can identify significantly larger numbers of DEGs in six libraries, thus providing more comprehensive genetic and genomic information.
Previous studies have reported that sclerotia are induced by reactive oxygen species (ROS) (Georgiou et al. 2000, 2006). O2·− radicals is formed from molecule oxygen (O2) by accepting a single electron. Subsequently, H2O2, which is toxic, is generated from O2·− radical in the presence of superoxide dismutase. However, H2O2 is an important intracellular messenger, which is stable and can cross cell membranes. When H2O2 captures an electron, a hydroxyl radical (OH) is generated and causes lipid peroxidation (Apel and Hirt 2004; Georgiou et al. 2006). Numerous studies have shown the dynamic progression of SD in fungi is accompanied by increasing levels of lipid peroxidation (Georgiou 2000; Georgiou and Petropoulou 2001). Since hyperoxidant states are harmful to cells, defense mechanisms within organisms have evolved to produce antioxidant molecules, such as glutathione, melanin, and ascorbic acid, and enzymes, including superoxide dismutase, catalase, and glutathione peroxidase, which neutralize the accumulation of ROS (Vijayakumar et al. 2009). In the present study, NADPH-related unigenes (TCONS_00009009, TCONS_00004890, and TCONS_00010386), as well as a glutathione reductase encoding gene (TCONS_00013972), were highly expressed at the MS and dramatically decreased at the SI and SM stages. Belonging to the disulfide reductase family, glutathione reductase can catalyze NADPH-dependent reduction of oxidized glutathione to reduced glutathione. The reduced environment of the cytosol is maintained by reduced glutathione and catalyzes oxidant xenobiotic molecules to form oxidized glutathione by glutathione S-transferase, to balance the GSSG/GSH redox state (Csiszár et al. 2016). We found that expression of glutathione S-transferase gene (TCONS_00013585) was high at the SM stage, as was the expression of the catalase gene (TCONS_00002674), which catalyzes the decomposition of H2O2 to water. Other stress regulators, such as heat shock proteins and oxidoreductase FAD/NAD-binding protein are also highly expressed at the SM stage. To protect the organism from harmful ROS and regulate the balance between cell survival and death, heat shock protein may be produced (Baruah et al. 2014). Overall, this study suggests that R. solani AG1-IA employs different pathways in response to oxidative stress.
During SD of R. solani AG1-IA, pigmented particles accumulate in intercellular spaces (Fig. 1). Melanin can protect an organism from UV light, ROS, and other adverse environmental factors; however, the biosynthetic pathways of melanin production are complex and involve numerous regulatory genes. Four different pathways have been previously identified in fungi, namely the1,8-dihydroxynaphthalene, 3, 4-dihydroxyphenylalanine, γ-glutaminyl-3,4-dihydroxybenzene, and catechol pathways (Chen et al. 2015). Considered the ubiquitous melanin biosynthetic pathway in Ascomycotina and Deuteromycotina and the biochemical basis of fungal 1,8-dihydroxynaphthalene melanin biosynthesis, the 1,8-dihydroxynaphthalene pathway has been well studied (Pal et al. 2014; Schumacher 2016; Upadhyay et al. 2013). However, the exact melanin biosynthetic pathway and related genes in R. solani AG1-IA are still unknown. In this study, three unigenes involved in the DHN biosynthesis of the pigment were observed, TCONS_0005501 (polyketide synthetase), TCONS_00008578 (polyketide cyclase), and TCONS_00006208 (laccase). Furthermore, one unigene (TCONS_00007083) encoding tyrosinase, which plays an important role in the DOPA pathway, was significantly increased at the SI stage. Laccase and tyrosinase not only participated in melanin synthesis, but also involved in the response to ROS. Hence, the upregulation of laccase and tyrosinase encoding genes at MS and SI may be due to the high level of ROS in these two stages. These results imply that the melanin synthesis pathway in R. solani AG1-IA may be more complicated than those in Verticillium dahliae, Sclerotinia sclerotiorum, and Exserohilum turcicum, which are well known DHN melanin-producing fungi. The exact functions of such highly expressed genes merit further investigations using heterologous expression method.
The morphological characteristics of R. solani AG1-IA SD have been well studied. In the SI stage, undifferentiated hyphae aggregate together to form monilioid cells. The masses of compact monilioid cells enlarge to form hyphal coils, which become spherical and white in color (Townsend and Willetts 1954). During SM, cells undergo autolysis and secrete a brown-colored fluid (Aliferis and Jabaji 2010). Morphogenesis from undifferentiated hyphae to sclerotia is thought to involve the molecular pathways of cellular reprogramming and including those involved in the cell cycle and growth, division and cytoskeleton, and autophagy and cell death (Duressa et al. 2013; Xiong et al. 2014). Our results support the possibility that increased expression of genes encoding for cell cycle activities may also be important for sclerotial formation. The plasma membrane fusion protein gene (TCONS_00014782), cellular morphogenesis protein gene (TCONS_00014075), and WD40 protein gene (TCONS_00001320) were upregulated in the SI library and may contribute to fungal development. Membrane fusion provides dynamic communication between membrane-bound organelles and is an important process in eukaryotes. Plasma membrane fusion protein is internalized from the plasma membrane and participates in endocytosis, which is likely to govern cell polarity and signal transduction (Higuchi et al. 2006). Recently, the WD40 domain protein has been widely studied, and plays key role in cellular networks, such as signal transduction, cell division, cytoskeleton assembly, chemotaxis, and RNA processing (Shim et al. 2006). Our results indicated that WD40 proteins not only involved in hyphal morphogenesis, energy metabolism, and conidiation, as well as pathogenicity (Yuan et al. 2017), but also sclerotial development in filamentous fungi. Furthermore, the actin cytoskeleton organization protein gene (TCONS_00013740) and cell cycle protein kinase spo4 gene (TCONS_00013288) showed the highest expression at the SM stage. Actin cytoskeleton organization is not only involved in cell morphology and polarity, it also governs the assembly and disassembly of filamentous actin structures. Moreover, actin cytoskeleton organization-related proteins are associated with cell wall morphogenesis and endocytosis, suggesting these proteins may play a role in regulating morphological transitions in SD.
The majority of signal transduction and energy metabolism pathways are involved in SD. During the process, an abundance of proteins are being made and degraded. Protein degradation plays an invaluable role in the physiological activity of cells, including the cell cycle, DNA reparation, cell growth and differentiation, control of cell quality, and apoptosis. The degradation of intracellular protein can be divided into two forms: autophagy and the ubiquitin-proteasome system (UPS). Autophagy plays an important role in degrading intracellular organelle and stable proteins, while UPS is responsible for the degradation of a vast majority of intracellular proteins (Korolchuk et al. 2009). Intriguingly, the unigenes involved in autophagy (TCONS_00011673, TCONS_00003071, TCONS_00013298, TCONS_00003033, and TCONS_00001409) showed consistently significant expression at the SI stage. Autophagy is a highly conserved cell degradation process in eukaryotes, and generally sustained at very low levels (Maiuri et al. 2007). However, autophagy is induced under stress conditions and autophagic activation can reduce ROS levels leading to acceleration of protein elimination (Li et al. 2015). In yeast, autophagy significantly increases during conditions of nutrient deficiency, which acts to degrade proteins no longer required, thereby recycling the amino acids necessary for synthetizing new proteins for survival (Kuma et al. 2004). Autophagy and UPS work synergistically in cells. Damaged proteins are modified by ubiquitin polymers, recognized by ubiquitin binding proteins, and transported to autohpagosomes for degradation. Interaction of ubiquitin binding proteins can improve the transport efficiency of autophagic vacuoles and increase autophagic flux (Geisler et al. 2010). By degrading the cyclin in cyclin-CDK complexes, UPS also regulates the cell cycle. In this study, the fact that the ubiquitin gene (TCONS_00003960) and ubiquitin-specific protease gene (TCONS_00008189) were highly expressed at the SM stage indicates that SM may involve complex protein degradation processes. The ubiquitin-protein ligase gene (TCONS_00008527) is highly expressed at the SI stage, and may play an important role in cell division and increase defenses against ROS.
In filamentous fungi, the progress of sclerotial development and maturation involves active translocation of molecules from hyphae to sclerotia. Some molecules are necessary for the biosynthesis of energy compounds. In this study, the increases in expression of genes for certain secondary metabolite biosynthetic proteins, carbohydrate metabolism proteins, transport proteins, and transcription factors are consistent with previous reports relating to V. dahliae (Xiong et al. 2014). While carbohydrate metabolism is involved in pathogenicity, it also plays a role in remodeling fungal cell walls during SM (Xiong et al. 2014). Three glycosyl hydrolase family proteins (TCONS_00004363, TCONS_00005255, and TCONS_00004868) and three polysaccharide lyase family proteins (TCONS_00005607, TCONS_00011017, and 00008054) were differentially regulated in the SI library, indicating they may be associated with energy metabolism during SD.
Our study surveyed three transcriptomes of the SD process in R. solani AG1-IA using RNA-Seq. Global gene expression patterns and stage-specific transcriptome changes during SD were demonstrated. Further analysis showed unigenes that are differentially regulated during SD are also involved in secondary metabolite biosynthesis, melanin biosynthesis, ubiquitin and autophagy, and ROS metabolism. Gene conservation survey showed that some DEGs were highly conserved in the genome of R. solani AG1-IB, R. solani AG3, and R. solani AG8. This showed SD-related genes were less variable in the complex species of Rhizoctonia. The sclerotium of R. solani AG1-IA is an important target for the control of RSB, and the unigenes annotated in this study will provide useful information for disease control strategies aimed at preventing the formation of sclerotia. Recently, transcriptome studies showed that some DEGs involved in the pathogenicity of R. solani (Chen et al. 2016), which provided useful information for the study of gene function. By comparative transcriptomic analysis, the results showed that SD relative genes were also involved in the pathogenicity progress. Some of them were highly expressed at the early stage of RSB, indicating that they may respond to pathogen recognition and colonization. The other DEGs showed high expression level at 32–72 h, indicating that they may play an important role in the disease progression. Based on this information, in our laboratory, we have screened several unigenes from this study using the virus-induced gene silencing method. The results showed that silencing of target genes (i.e., TCONS_000113585) confers resistance of Nicotiana benthamiana to R. solani AG1-IA when challenge inoculation with this isolate (unpublished data). We will create resistant new cultivars of rice which show enhanced resistance to rice sheath blight using host-induced gene silencing techniques in the future.
Taken together, this study has laid the foundation for the further application of functional genomics in R. solani AG1-IA.
References
Aliferis KA, Jabaji S (2010) 1 H NMR and GC-MS metabolic fingerprinting of developmental stages of Rhizoctonia solani sclerotia. Metabolomics 6:96–108
Altschul SF, Gish W, Miller W, Myers E, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55:373–399
Baruah K, Norouzitallab P, Linayati L, Sorgeloos P, Bossier P (2014) Reactive oxygen species generated by a heat shock protein (Hsp) inducing product contributes to Hsp70 production and Hsp70-mediated protective immunity in Artemia franciscana against pathogenic vibrios. Dev Comp Immunol 46:470–479
Brown MF, Wyllie TD (1970) Ultrastructure of microsclerotia of Verticillium alboatrum. Phytopathology 52:538–542
Chen XY, Wang W, Wang ZY, Chen WL (2012) Analysis of differentially expressed genes of Rhizoctonia solani during the sclertium formation. J Zheijang Univ 38:28–34
Chen J, Wang C, Shu C, Zhu M, Zhou E (2015) Isolation and characterization of a melanin from Rhizoctonia solani, the causal agent of rice sheath blight. Eur J Plant Pathol 142:281–290
Chen L, Ai P, Zhang J, Deng Q, Wang S, Li S, Zhu J, Li P, Zheng A (2016) RSIADB, a collective resource for genome and transcriptome analyses in Rhizoctonia solani AG1 IA. Database 2016:w31
Csiszár J, Horváth E, Bela K, Gallé Á (2016) Glutathione-related enzyme system: glutathione reductase (GR), glutathione transferases (GSTs) and glutathione peroxidases (GPXs). Springer International Publishing
Daly P, van Munster JM, Kokolski M, Sang F, Blythe MJ, Malla S, Oliveira JV, Goldman GH, Archer DB (2016) Transcriptomic responses of mixed cultures of ascomycete fungi to lignocellulose using dual RNA-seq reveal inter-species antagonism and limited beneficial effects on CAZyme expression. Fungal Genet Biol 102:4–21
Damicone JP, Patel MV, Moore WF (1993) Density of sclerotia of Rhizoctonia solani and incidence of sheath blight in rice fields in Mississippi. Plant Dis 77:257–260
Duressa D, Anchieta A, Chen D, Klimes A, Garcia-Pedrajas MD, Dobinson KF, Klosterman SJ (2013) RNA-seq analyses of gene expression in the microsclerotia of Verticillium dahliae. BMC Genomics 14:607
Feng S, Shu C, Wang C, Jiang S, Zhou E (2017) Survival of Rhizoctonia solani AG-1 IA, the causal agent of rice sheath blight, under different environmental conditions. J Phytopathol 165:44–52
Finotello F, Di CB (2015) Measuring differential gene expression with RNA-seq: challenges and strategies for data analysis. Brief Funct Genomics 14:130–142
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131
Georgiou CD (2000) Lipid peroxidation in Sclerotium rolfsii: a new look into the mechanism of sclerotial biogenesis in fungi. Mycol Res 101:460–464
Georgiou CD, Petropoulou KP (2001) Effect of the antioxidant ascorbic acid on sclerotial differentiation in Rhizoctonia solani. Plant Pathol 50:594–600
Georgiou CD, Tairis N, Sotiropoulou A (2000) Hydroxyl radical scavengers inhibit sclerotial differentiation and growth in Sclerotinia sclerotiorum and Rhizoctonia solani. Mycol Res 104:1191–1196
Georgiou CD, Patsoukis N, Papapostolou I, Zervoudakis G (2006) Sclerotial metamorphosis in filamentous fungi is induced by oxidative stress. Integr Comp Biol 46:691–712
Higuchi Y, Nakahama T, Shoji JY, Arioka M, Kitamoto K (2006) Visualization of the endocytic pathway in the filamentous fungus Aspergillus oryzae using an EGFP-fused plasma membrane protein. Biochem Biophys Res Commun 340:784–791
Hu W, Pan X, Abbas HMK, Li F, Dong W (2017) Metabolites contributing to Rhizoctonia solani AG-1-IA maturation and sclerotial differentiation revealed by UPLC-QTOF-MS metabolomics. PLoS One 12(5):e0177464
Korolchuk VI, Menzies FM, Rubinsztein DC (2009) A novel link between autophagy and the ubiquitin-proteasome system. Autophagy 5:862–863
Kuanar SR, Ray A, Sethi SK, Chattopadhyay K, Sarkar RK (2017) Physiological basis of stagnant flooding tolerance in rice. Rice Science 24(2):73–84
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432:1032–1036
Kwon YS, Kim SG, Chung WS, Bae H, Jeong SW, Shin SC, Jeong MJ, Park SC, Kwak YS, Bae DW (2014) Proteomic analysis of Rhizoctonia solani AG-1 sclerotia maturation. Fungal Biol 118:433–443
Li L, Jin T, Miao Y, Ping L, Qiang Z (2015) ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol 35:615–621
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔ CT method. Methods 25:402–408
Lu L, Shu C, Liu C, Wang C, Zhou E (2016) The impacts of natural antioxidants on sclerotial differentiation and development in Rhizoctonia solani AG-1 IA. Eur J Plant Pathol 146:729–740
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–752
Maurya S, Singh UP, Singh R, Singh A, Singh HB (2010) Role of air and light in sclerotial development and basidiospore formation in Sclerotium rolfsii. J Plant Res 50:206–209
Moni ZR, Ali MA, Alam MS, Rahman MA, Bhuiyan MR, Mian MS, Iftekharuddaula KM, Latif MA, Khan MAI (2016) Morphological and genetical variability among Rhizoctonia solani isolates causing sheath blight disease of rice. Rice Science 23(1):42–50
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
New CM, Coley-Smith JR, Georgy NI (1984) Scanning electron microscopy of sclerotial germination in Sclerotium cepivorum. Trans Br Mycol Soc 83:690–693
Pal AK, Gajjar DU, Vasavada AR (2014) DOPA and DHN pathway orchestrate melanin synthesis in Aspergillus species. Med Mycol 52:10–18
Ritchie F, Bain RA, Mcquilken MP (2009) Effects of nutrient status, temperature and pH on mycelial growth, sclerotial production and germination of Rhizoctonia solani from potato. J Plant Pathol 91:589–596
Sarrocco S, Mikkelsen L, Vergara M, Dan FJ, Lübeck M, Vannacci G (2006) Histopathological studies of sclerotia of phytopathogenic fungi parasitized by a GFP transformed Trichoderma virens antagonistic strain. Mycol Res 110:179–187
Schumacher J (2016) DHN melanin biosynthesis in the plant pathogenic fungus Botrytis cinerea is based on two developmentally regulated key enzyme (PKS)-encoding genes. Mol Microbiol 99:729–748
Shim WB, Sagaram US, Choi YE, So J, Wilkinson HH, Lee YW (2006) FSR1 is essential for virulence and female fertility in Fusarium verticillioides and F. graminearum. Molecular plant-microbe interactions. MPMI 19:725–733
Shu C, Chen J, Sun S, Zhang M, Wang C, Zhou E (2015) Two distinct classes of protein related to GTB and RRM are critical in the sclerotial metamorphosis process of Rhizoctonia solani AG-1 IA. Funct Integr Genomics 15:449–459
Srinivas P, Babu SR, Ratan V (2014) Role of sclerotia, plant debris and different hosts on survival of rice sheath blight pathogen, Rhizoctonia solani. Int J Appl Biol Pharm Biotechnol 5:29–33
Sumner DR (1996) Sclerotia formation by Rhizoctonia species and their survival. Springer, Netherlands
Taheri P, Tarighi S (2011) Cytomolecular aspects of rice sheath blight caused by Rhizoctonia solani. Eur J Plant Pathol 129:511–528
Townsend BB, Willetts HJ (1954) The development of sclerotia of certain fungi. Trans Br Mycol Soc 37:213–221
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578
Upadhyay S, Torres G, Lin X (2013) Laccases involved in the DHN melanin biosynthesis in Aspergillus fumigatus are regulated by developmental factors and copper homeostasis. Eukaryot Cell 12:1641–1652
Vijayakumar R, Zhao CX, Gopal R, Jaleel CA (2009) Non-enzymatic and enzymatic antioxidant variations in tender and mature leaves of Strychnos nux-vomica L. (Family: Loganiaceae). CR Biol 332:52–57
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang ZX, Zhou XZ, Meng HM, Liu YJ, Zhou Q, Huang B (2014) Comparative transcriptomic analysis of the heat stress response in the filamentous fungus Metarhizium anisopliae using RNA-Seq. Appl Microbiol Biotechnol 98:5589–5597
Wang L, Liu LM, Hou YX, Li L, Huang SW (2015) Pathotypic and genetic diversity in the population of Rhizoctonia solani AG1-IA causing rice sheath blight in China. Plant Pathol 64:718–728
Wang YN, Lei T, Yan H, Ping W, Hua Y, Wei CL (2016) Differential transcriptome analysis of leaves of tea plant (Camellia sinensis) provides comprehensive insights into the defense responses to Ectropis oblique attack using RNA-Seq. Funct Integr Genomics 16:383–398
White JG (1976) Effects of temperature, light and aeration on the production of microsclerotia by Pyrenochaeta lycopersici. Trans Br Mycol Soc 67:497–498
Willetts HJ, Bullock S (1992) Developmental biology of sclerotia. Mycol Res 96:801–816
Xiong D, Wang Y, Ma J, Klosterman SJ, Xiao S, Tian C (2014) Deep mRNA sequencing reveals stage-specific transcriptome alterations during microsclerotia development in the smoke tree vascular wilt pathogen, Verticillium dahliae. BMC Genomics 15:324
Xu L, Zhu L, Tu L, Liu L, Yuan D, Jin L, Long L, Zhang X (2011) Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. J Exp Bot 62:5607–5621
Yuan L, Su Y, Zhou S, Feng Y, Guo W, Wang X (2017) A RACK1-like protein regulates hyphal morphogenesis, root entry and in vivo virulence in Verticillium dahliae. Fungal Genet Biol 99:52–61
Zheng A, Lin R, Zhang D, Qin P, Xu L, Peng A, Lei D, Wang Y, Yao C, Yao L (2013) The evolution and pathogenic mechanisms of the rice sheath blight pathogen. Nat Commun 4:1424
Acknowledgments
This work was supported by a grant from a National Natural Science Foundation of China (Grant No. 31801677) awarded to Canwei Shu, and a Natural Science Foundation of Guangdong Province for Doctoral program (Grant No. 2016A030310454) awarded to Canwei Shu and the National Natural Science Foundation of China (Grant Nos. 31271994 and 31470247) awarded to Erxun Zhou.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Fig. S1
Length distribution of assembled unigenes. (PNG 47 kb)
Fig. S2
Length distribution of coding sequences (CDS) produced by searching unigenes against Nr database using BLAST2GO (E-value <10−5) and size distribution of the proteins predicted from the CDS sequences. (PNG 79 kb)
Fig. S3
Short time-series expression miner (STEM) clustering of DEGs. Eight profiles ordered based on significance (p value) of the number of genes assigned versus expected. Profile 4 (0.0, 0.0, 1.0): 1218 genes assigned, 1069.8 genes expected, p = 1.2E−06 (significant). Profile 1 (0.0,-1.0,-1.0): 1210 genes assigned, 1069.8 genes expected, p = 4.1E−06 (significant). Profile 0 (0.0,-1.0,-2.0): 877 genes assigned, 761.0 genes expected, p = 9.5E−06 (significant). Profile 2 (0.0,-1.0, 0.0): 1828 genes assigned, 1710.7 genes expected, p = 0.00097 (significant). Profile 7 (0.0, 1.0, 2.0): 836 genes assigned, 761.0 genes expected, p = 0.0027 (significant). Profile 3 (0.0, 0.0,-1.0): 1217 genes assigned, 1308.5 genes expected, p = 1 (not significant). Profile 5 (0.0, 1.0, 0.0): 1048 genes assigned, 1428.7 genes expected, p = 1 (not significant). Profile 6 (0.0, 1.0, 1.0): 1184 genes assigned, 1308.5 genes expected, p = 1 (not significant). (PNG 93 kb)
Fig. S4
Venn diagram illustrating number of DEGs co-existed in different Rhizoctonia solani AGs (i.e., R. solani AG1-IB, R. solani AG3 and R. solani AG8). (PNG 30 kb)
Table S1
(DOCX 14 kb)
Table S2
(DOCX 16 kb)
Table S3
(DOCX 14 kb)
Rights and permissions
About this article

Cite this article
Shu, C., Zhao, M., Anderson, J.P. et al. Transcriptome analysis reveals molecular mechanisms of sclerotial development in the rice sheath blight pathogen Rhizoctonia solani AG1-IA. Funct Integr Genomics 19, 743–758 (2019). https://doi.org/10.1007/s10142-019-00677-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-019-00677-0